Article Text

Abstract
OBJECTIVE To establish the pathophysiological mechanisms of striatopallidal and thalamic dystonia.
METHODS Five patients from among 26 who presented (between March 1987 and July 1996) with focal dystonia, segmental dystonia, or hemidystonia caused by a single localised vascular lesion, were selected. Patients with lesions with indefinite boundaries, and diffuse, or multiple, or large brain lesions were excluded. Three dimensional T1 weighted MRI (1.5 tesla) was performed to determine the topography of the lesions. The atlas of Hassler allowed the stereotactic localisation of the lesions to be specified exactly.
RESULTS Three patients had dystonic spasms associated with striatopallidal lesions and one with a thalamic and striatopallidal lesion. One other patient presented with a myoclonic dystonia related to a thalamic lesion. The striatopallidal lesions were located in the sensorimotor area with a somatotopical distribution. The pure thalamic lesion involved the centromedian nucleus, the sensory nuclei, and the pulvinar whereas the thalamic and striatopallidal lesion was located in the pallidonigral thalamic territory, which receives pallidonigral inputs.
CONCLUSION The striatopallidal dystonia might be the consequence of the interruption of the cortico-striato-pallido-thalamo-cortical loop induced by lesions located within the sensorimotor part of the striatopallidal complex. By contrast, it is suggested that thalamic dystonia might be caused by lesions located in the centromedian or the ventral intermediate nuclei, outside the pallidonigral territory, but leading also to a dysfunction of the cort ico - striat o - pallido - thalamo - cort ica l loop.
- dystonia
- basal ganglia
- thalamus
- stroke
Statistics from Altmetric.com
Focal brain lesions can induce several types of abnormal involuntary movements—for example, dystonia, chorea, hemiballism, tremor, myoclonus, parkinsonism, and asterixis.1 2 The most frequent cause is stroke, followed by tumour, trauma, anoxia, vascular malformation, or multiple sclerosis.1 2 For some causes such as stroke or anoxia these abnormal involuntary movements can occur immediately after the brain lesion (for example, hemiballism),3 when the initial motor deficit improves or recovers, or after a long period (a few months or years) of stability of the motor impairment.4 5 Among these abnormal involuntary movements, dystonia, which is particularly common, is often related to vascular lesions, usually located within the striatopallidal complex,4 6-11 the thalamus,4 10 12–14 or the brainstem.2Most of the striatopallidal lesions are located in the lentiform nucleus, especially the putamen.4 With thalamic lesions, myoclonic dystonia is not commonly reported.10 15 Stroke can induce unique localised lesions with well defined boundaries that facilitate anatomical-clinical correlations but only a few neuropathological studies have been published.6 12 16Furthermore it remains difficult to determine in vivo the exact localisation of the lesions because of the lack of precision of conventional radiology; indeed, most of the studies used CT and standard MRI, which could not specify exactly which striatopallidal area—sensorimotor or associative—was involved, or to what extent there was a somatotopical distribution of the lesion, or which thalamic nuclei were affected.1 2 Only Lehéricy et al has used methods similar to ours,10 one which allows a more precise approach to the location of lesions.
The aim of this study was to establish the pathophysiological mechanisms of striatopallidal and thalamic dystonia by establishing precise clinicoradiological correlations. This was facilitated by our recent knowledge on the circuitry of the basal ganglia.17We chose therefore to study stroke patients with focal lesions. They underwent three dimensional T1 weighted MRI sequence (1.5 tesla) to determine the precise topography of lesions by stereotactic analysis using the atlas of Hassler.18 We then compared the lesions with clinical findings to clarify pathophysiological mechanisms.
Patients and methods
PATIENTS
Patients were selected according to the following criteria: all of them had presented with a stroke (infarct or haemorrhage) initially responsible for hemiplegia, followed by symptomatic dystonic spasms or myoclonic dystonia of delayed onset. Dystonic spasms were defined as sustained muscle contractions responsible for twisting and repetitive movements or abnormal postures, and were triggered by postures and increased by voluntary movements and gait, whereas they were absent at rest.19 Myoclonic dystonia consisted of sustained muscle contractions overlain by myoclonic jerks.20 Symptomatic dystonia was defined according to the following criteria: unilateral dystonia (contralateral to the vascular lesion), presence of additional neurological abnormalities, rapidly progressive evolution, and unusual course in terms of the distribution of parts of the body affected or age at onset.21 Dystonia affected the ipsilateral arm and leg (hemidystonia), two or more contiguous parts of the body (segmental dystonia), or a single part of the body (focal dystonia).19 According to CT and standard MRI, the lesions were located within the basal ganglia,or the thalamus, or both. Thus 26 consecutive patients were first retrospectively selected among patients admitted to the neurology departments of R Salengro Hospital, Lille between March 1987 and July 1996. Of this first group, four patients were lost to follow up. Two patients refused another MRI. Thirteen patients were excluded because of lesions with indefinite boundaries, or multiple or large brain lesions not suitable for precise radiological analysis.
Finally, seven patients had head MRI according to methods described later. Two of these seven patients were excluded because of a lesion with indefinite boundaries in one and artefacts due to abnormal involuntary movements in the second. The MRI of the five remaining patients—with a single localised vascular lesion—was analysed using Hassler’s atlas.18
METHODS
Imaging was performed with a 1.5 tesla MR unit (Siemens Vision, Erlangen) and standard head coil. A 3D T1 weighted MP-RAGE sequence (TR=9.7 ms; TE=4 ms; TI=300 ms; field of view=256 mm; matrix=256×256; two acquisitions) was used, allowing 1 mm isometric pixel size. After acquisition, data were analysed and reconstructed on a Siemens workstation using Numaris V 4.3 software. The bicommissural plane on the midsagittal slice was identified and 1 mm contiguous axial slices were reconstructed. Afterwards, sagittal and coronal planes were defined from the bicommissural plane and 1 mm contiguous slices were reconstructed. On magnified images, bicommissural distance was defined as the shortest distance between opposing surfaces of the commissures.18 The middle of the bicommissural line was used to specify three dimensional lesion coordinates for each plane and slice. Then, measures were manually reported on Hassler’s atlas according to bicommissural length, width of the third ventricle, and thalamus size.18 Boundaries of each lesion were reconstructed on Hassler’s atlas on axial, coronal, and sagittal planes to determine the different structures involved and to assess the lesion volume. Thalamus segmentation was studied according to the classification of Hirai and Jones, which is commonly used and provides a good definition of thalamic physiological territories.22
Results
CLINICAL FINDINGS
Clinical features of the patients (four women and one man), all right handed, are summarised in table 1. Mean age at the time of the study was 55.8 years. Causes of dystonia were either haemorrhage (patient 3), or infarcts (other patients). The interval between stroke and onset of dystonia varied from 6 weeks (patient 1) to 7 months (patient 5) (mean 3.2 months). Dystonia occurred when patients had almost or completely recovered from hemiplegia.
Clinical features
Patients 1, 2, 4, and 5 presented with dystonic spasms whereas patient 3 presented with myoclonic dystonia. There were two cases of hemidystonia (patients 1 and 2), one case of segmental dystonia (patient 5), and two cases of focal dystonia (patient 3, hand; patient 4, foot). Patient 1 had extension of the fingers, pronation of the forearm, and sometimes abduction and external rotation of the arm. There was also a hyperextension of the big toe alternating with a flexion of the other toes and equine varus attitude of the foot. Patient 2 had internal rotation of the arm, hyperextension of the wrist and of the metacarpophalangeal joints, adduction of the thumb and the hand, and equinovarus foot. Patient 4 had hyperextension of the big toe and adduction of the second and third toes. Patient 5 had dystonic deep hand, flexion of the fingers, and pronation of the forearm. Patient 3 had flexion and pronation of the hand and superimposed distal and proximal myoclonic jerks.
At the time of the study, patients 2, 3, 4, and 5 had sequelae from stroke: mild hemiparesis with Babinski’s sign and hyperreflexia (patient 2 and 3), tactile hemihypoaesthesia, (patient 4) and spastic hypertonia of the lower limb with hyperreflexia (patient 5).
IMAGING RESULTS
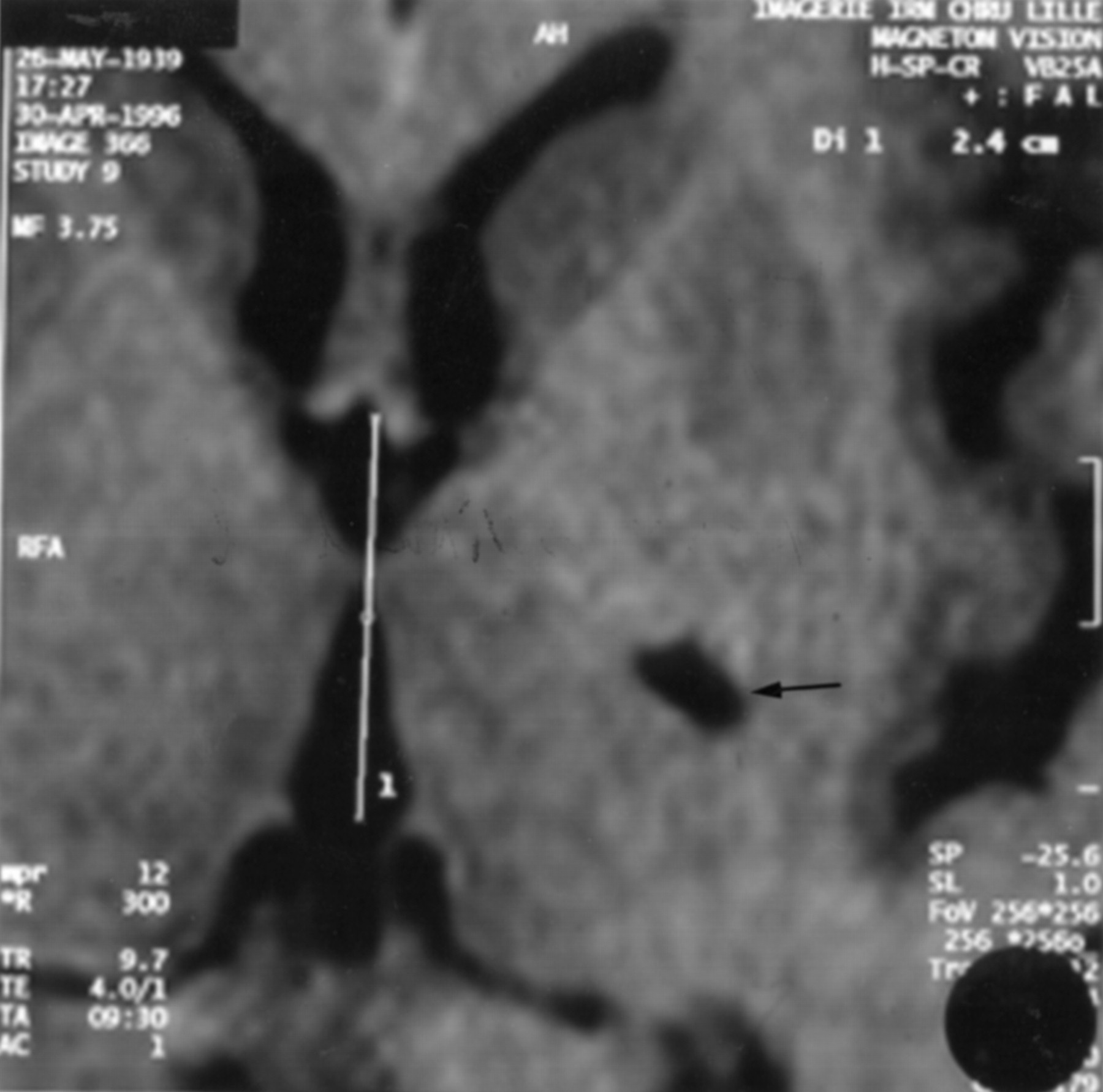
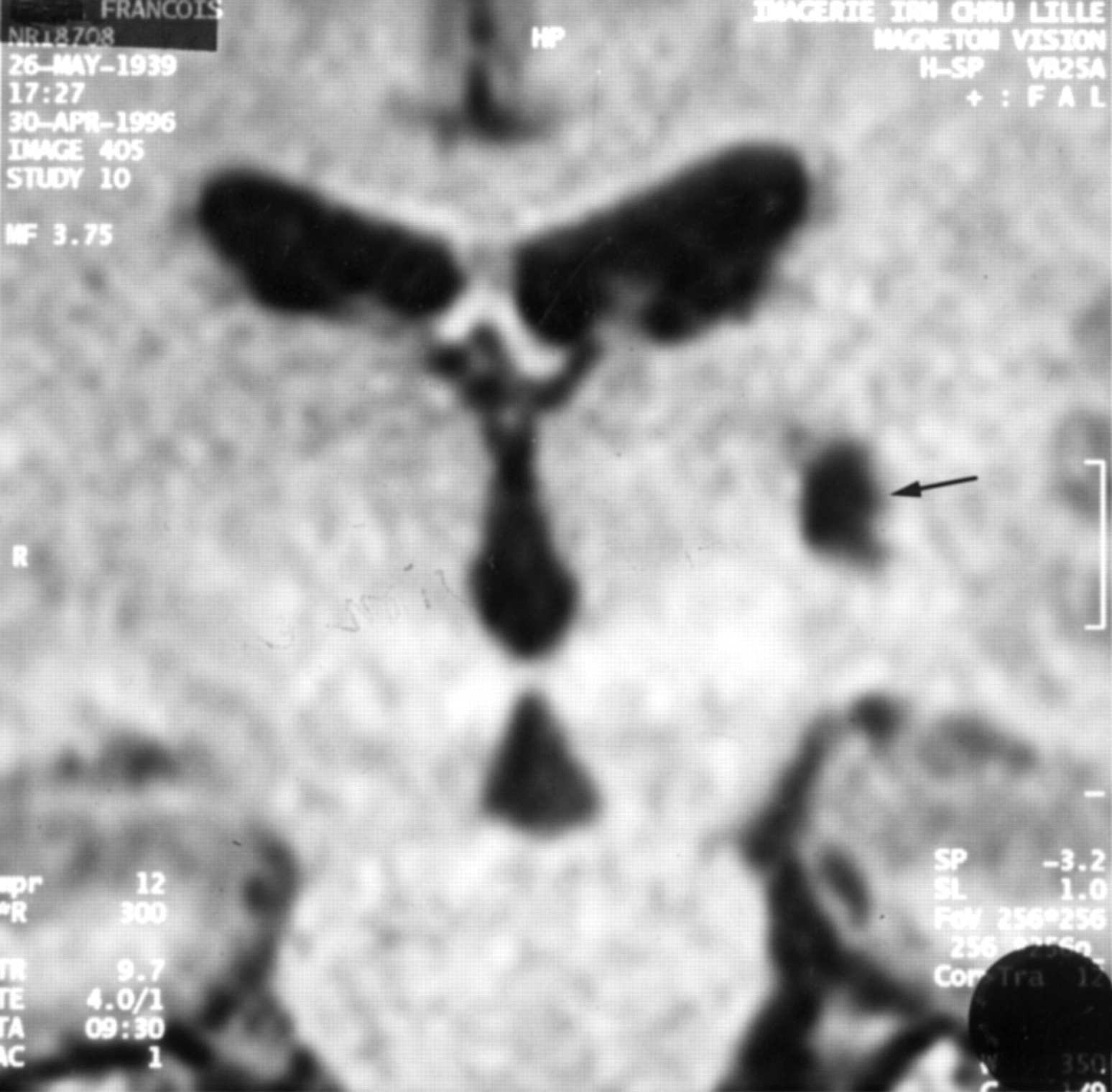
Imaging results are summarised in table 2 and are illustrated from patient 5 in figs 1-4; figs 1 and 2 show respectively the axial reconstruction according to the bicommissural line and the report on Hassler’s atlas and figs 3 and 4 show the coronal reconstruction and the report on the atlas.18 Estimated volume of lesions varied from 88 mm3 to 2863 mm3 (mean 1032 mm3). Lesions were hypointense on T1 weighted images, which provided good delineation of boundaries. Bicommissural distance (23 to 29 mm; mean, 25.2 mm) was in accordance with the measures of Schaltenbrand performed on 111 brains for the atlas.18 Three patients (1, 4, and 5) had lesions located exclusively within the striatopallidal complex. Patient 2 presented with both thalamic and striatopallidal lesions. Patient 3 had a pure thalamic lesion. The postcommissural part of the putamen was predominantly affected (except patient 1: precommissural and postcommissural putamen), as was the head and body of the caudate nucleus (except patient 5). In two patients (1 and 4), the lesions were located in the putamen with a somatotopical distribution; for the segmental dystonia of the upper limb (patient 4) and the hemidystonia (patient 1), putaminal lesions involved the somatotopic area corresponding respectively to the upper limb and both upper and lower limbs.23 The internal pallidum and external pallidum were similarly involved in patients 2 and 5 whereas only the external pallidum was affected in patients 1 and 4. The thalamic lesion in patient 2 involved a part of the pallidonigral territory (ventral anterior nucleus; ventral lateral anterior nucleus). For the pure thalamic lesion (patient 3), the centromedian-parafascicularis nucleus, the pulvinar, and the sensory nuclei (ventral posterior lateral nucleus; ventral posterior medial nucleus; ventral medial basal nucleus) were affected.
Imaging results (the lesions are classified for each structure involved according to the ratio (%) between the involved volume and the volume taken by the structure on the atlas of Hassler10)

Three dimensional T1weighted MRI sequence (MP-RAGE): axial reconstruction according to the bicommissural line, showing the lenticular lesion; the bicommissural line is superimposed on the slice.

Lesion coordinates (axial reconstruction) are superimposed on the atlas of Hassler. C.c=corpus callosum; Cd=caudate nucleus; Cl=claustrum; Cp.e=external capsule; Cp.i=internal capsule; Cx.in=insular cortex; Fx=fornix; La.p.l=lamina pallidi lateralis; P.l=globus pallidus externus; Put=putamen; Th=thalamus; Ve.l=lateral ventricle.

Three dimensional T1 weighted MRI sequence (MP-RAGE): coronal reconstruction according to the bicommissural line, showing the lenticular lesion.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lesion coordinates (coronal reconstruction) are superimposed on the atlas of Hassler. C.c=corpus callosum; Cd=caudate nucleus; Cp.e=external capsule; Cp.i=internal capsule; Fx=fornix; HF=hippocampal formation; La.p.i=lamina pallidi incompleta; La.p.l=lamina pallidi lateralis; La.p.m=lamina pallidi medialis; Ni=substantia nigra; P.l=globus pallidus externus; P.m.e=globus pallidus internus pars lateralis; P.m.i=globus pallidus internus pars medialis; Ps.pd=cerebral peduncle, pars superior; Put=putamen; Th=thalamus; Sth=subthalamic nucleus; Ve.l=lateral ventricle.
Discussion
Our study has shown that a three dimensional T1 weighted MRI associated with a stereotactic analysis using Hassler’s atlas,18 provided precise clinicoradiological correlations. Our MRI technique is clearly better than CT and standard MRI. It allows excellent contrast so that valuable delineation of the basal ganglia is possible, and provides precise localisation of the lesions despite some factors which limit its precision (an order of mm).24-26 Indeed, reconstructions based on Hassler’s atlas can pose difficulties regarding the localisation of the lesions: (1) the atlas is based on a single brain for each plane, and does not take into account anatomical variations; (2) the interslice space varies according to the situation of the slice in relation to the centre of the bicommissural plane, and furthermore, the axial plane cannot be used because of an angle of 7° with the bicommissural plane27; (3) our method provided only rigid transformations, which consisted of translation or rotation of reconstructed boundaries, so that complicated deformations could not be taken into consideration.27 28 Computerised methods using elastic deformations would have given better results.28-31 However, this problem did not influence our study because these patients had small lesions inducing only moderate displacements.
For the patients with striatopallidal lesions, patients 2, 4, and 5 had lesions located within the postcommissural putamen, which is included in the sensorimotor part of the striatum, whereas patient 1 had a lesion located within the postcommissural and precommissural putamen, which is included in the associative part of the striatum.32 Moreover, the lateral and ventral part of the head of the caudate nucleus, which is a sensorimotor part of the striatum,33 was affected in patients 1, 2, and 4. Our results, and suggestions by others,10 34 indicate that the sensorimotor part of the striatum plays an important part in the induction of dystonia. However, cases of dystonia induced by pure lesions of the caudate nucleus are rare,4 35 and we think that this may be due to the fact that the sensorimotor area of the caudate nucleus remains unaffected in most cases because of its very restricted size. The findings from patients 1 and 4 suggested a somatotopical distribution within the sensorimotor putamen, also mentioned in an earlier report.10 For patients 2 and 5, the lesion was somatotopically located in the face area of the putamen23; however, given the fact that dystonia involved both the upper and lower limbs in patient 2, and the upper limb alone in patient 5, this could possibly be due to the somatotopical organisation of the pallidal lesion. We were unable to determine if the sensorimotor part of the pallidum was also predominantly affected (with or without a somatotopical distribution) in our patients as the sensorimotor and associative areas in the pallidum are usually less well defined than in the putamen.33
As has already been suggested,1 4 putaminal lesions might interrupt both direct and indirect pathways.36 The underactivity of the indirect pathway might predominate and this disruption could increase the thalamocortical drive and induce dystonia. The experiments of Mitchell et al are partially in agreement with this hypothesis: there was underactivity of the indirect pathway but increased activity of the direct pathway.37 Both hypotheses lead to the same conclusion: dystonia could be induced by an increased thalamocortical drive. In cases of putaminal lesions, there could be an interruption of the cortico-striato-pallido-thalamo-cortical loop. This is backed up by a recent PET study in acquired hemidystonia due to striatopallidal lesions38: there was an increased activity in cortical motor areas, both receiving inputs from the pallidonigral thalamic territory. This is in agreement with our four patients, who had lesions in the lentiform nuclei and especially in the putamen (patients 1 and 4). The pallidal involvement (especially the external pallidum, as the internal globus pallidum was spared in patients 1 and 4) in our three patients (1, 4, 5) could also play a part in the induction of dystonia, as has been reported in humans and monkeys.1 39 40 We suggest that dystonia can be induced by increased thalamocortical drive due to an interruption of the cortico-striato-pallido-thalamo-cortical loop by lesions located in the sensorimotor part of the striatopallidal complex and not by putaminal lesions alone.
In patient 2, we think that the thalamic lesion, located within the pallidonigral territory (ventral nucleus, ventral lateral anterior nucleus),22 41 cannot induce dystonia for several reasons: firstly, if we take into account the increased thalamocortical drive hypothesis, a lesion of the thalamic pallidonigral territory should suppress dystonia by an inhibition of the thalamocortical drive overactivity rather than inducing it, although paradoxically the surgical treatment of dystonia uses a stereotactic target located outside the pallidonigral territory (thalamotomy or chronic thalamic stimulation).42-44 Also, to our knowledge, there is no reported case of thalamic dystonia associated with a vascular lesion located within the polar artery territory (including the pallidonigral territory). This may suggest that striatopallidal dystonia and thalamic dystonia have different pathophysiological bases, although both induce overactivity in motor areas.38 Thalamic dystonia would be associated with lesions located outside the pallidonigral territory.
In patient 3 with a pure thalamic lesion, the pallidonigral territory was spared,22 41 as also noted in the patients described by Lehéricy et al.10 This suggests that physiopathology of dystonia in these cases is different from that with striatopallidal lesions. A large part of the lesion was located in the centromedical nucleus (CM) (essential regulator of the cortico-striato-pallido-thalamo-cortical loop),36 as in the four patients described by Lehéricy et al.10 We think that it could play an important part in the induction of dystonia. This hypothesis is supported by experiments which suggest an inhibiting role of the mesial thalamus (including the CM) on the ventral lateral nucleus, which receives pallidal inputs.45 A PET study, in which overactivity was seen in motor areas after thalamic lesions,38 is in agreement with the increased thalamocortical drive hypothesis. Moreover, the experiments of Mitchell et al suggest that the CM could also induce dystonia by lesion of the inputs from the pedunculopontine complex.37 Given topographical errors due to methods (methods allow 1 mm thin sections minimising partial voluming and multiplanar reconstruction),10 and given involvement of sensory nuclei,22 the ventral intermediate nucleus of Hassler18 (ventral part of ventral lateral posterior nucleus of Hirai and Jones22), which is very close to the ventral posterior nucleus, could also be affected and play a part in the induction of dystonia although it was seemingly spared: the ventral intermediate nucleus was involved in all patients of Lehéricy et al 10; dystonia can also be induced by lesions of the cerebellar pathways which pass through the ventral intermediate nucleus,46 as has already been reported in humans,47-50 and animal experiments.51-53 Finally, the primary and accessory motor areas receive inputs from both thalamic pallidal and thalamic cerebellar territories,33 and a PET study in acquired hemidystonia induced by thalamic lesions disclosed an increased activity in motor areas.38 The pulvinar was affected, as in two patients (5 and 6) described by Lehéricy et al,10 but was not involved in patients 7 and 8 of their series. Thus, it seems unlikely that this nucleus is responsible for dystonia. The last nuclei affected were the sensory nuclei (ventral posterior lateral nucleus, ventral posterior medial nucleus, ventral medial basal nucleus) also mentioned by Lehéricy et al.10 However, the PET study mentioned above did not display variations of activity in cortical sensory areas (receiving inputs from the thalamic sensory territory).38 Thus it is unlikely that the sensory nuclei can induce dystonia.
Therefore, thalamic dystonia and striatopallidal dystonia might not share the same physiopathological bases but might both lead to a dysfunction of the cortico-striato-pallido-thalamo-cortical loop leading to an overactivity in primary and accessory motor areas. Whether an isolated lesion of the CM or the ventral intermediate nucleus, or a lesion affecting both, can induce dystonia, remains unknown. Furthermore, we still do not know why lesions located in similar areas may not necessarly cause dystonia.