Article Text

Abstract
Morvan’s fibrillary chorea is a rare disease characterised by symptoms which include neuromyotonia, cramping, weakness, pruritis, hyperhidrosis, insomnia, and delirium. The first case of Morvan’s fibrillary chorea to be associated with clinical manifestations of myasthenia gravis with thymoma, psoriasis, and atopic dermatitis is reported. Muscle histopathology disclosed chronic denervation and myopathic changes and in vitro electrophysiology demonstrated both presynaptic and postsynaptic defects in neuromuscular transmission. Serum antibodies to acetylcholine receptors, titin, N-type calcium channels, and voltage gated potassium channels were detected. Plasmapheresis, thymectomy, and long term immunosuppression induced a dramatic resolution of symptoms. The association of thymoma with other autoimmune disorders and autoantibodies, and prolonged and sustained remission with chronic immunosuppression, place Morvan’s fibrillary chorea on the range of neurological diseases arising as a paraneoplastic complication of cortical thymomas.
- Morvan’s fibrillary chorea
- neuromyotonia
- thymoma
- paraneoplastic
Statistics from Altmetric.com
La chorée fibrillaire was first coined by Morvan in 1890 when describing patients with multiple, irregular contractions of the long muscles, cramping, weakness, pruritis, hyperhidrosis, insomnia, and delirium.1 Additional cases of Morvan’s fibrillary chorea have been reported, but only four cases are in the English literature, perhaps as a matter of semantics.2 3 Neuromyotonia and Isaacs’ syndrome (continuous muscle fibre activity) are used interchangeably to refer to muscle twitching and cramping at rest, that is exacerbated with exercise.4 It is caused by sustained or repetitive spontaneous muscle activity of peripheral nerve origin. Electromyography discloses spontaneous, repetitive motor unit or single fibre discharges firing in irregular rhythmic bursts at high intraburst frequencies. Myokymia, or spontaneous rippling and twitching movements of muscles, is a visible component of neuromyotonia and Isaacs’ syndrome.5 Morvan’s fibrillary chorea is a syndrome that is characterised by not only neuromyotonia, but also dysautonomia and CNS dysfunction, often in the form of encephalopathy.
The association of the disease with thymoma, tumour, autoimmune diseases, and autoantibodies suggests an autoimmune or paraneoplastic aetiology. Thymoma, prostate adenoma, and in situ carcinoma of the sigmoid colon, have been found in patients with Morvan’s fibrillary chorea.2 3 In other patients, heavy metal intoxication with gold, mercury, and manganese, known stimulants of the immune system, has been implicated as a cause.6-8Anti-acetylcholine receptor (anti-AChR) antibodies have been detected in patients with thymoma, but without clinical manifestations of myasthenia gravis.2 Raised CSF IgG concentrations and oligoclonal bands have been reported in patients with psychosis.3 9
In addition, some of the individual components of Morvan’s fibrillary chorea are suspected of having an autoimmune or paraneoplastic basis because of the diseases and autoantibodies which are associated with them. Neuromyotonia, the most consistent component of Morvan’s fibrillary chorea, has been associated with myasthenia gravis, thymoma, humorally mediated peripheral neuropathies, neoplasm, and various autoantibodies.4 Recently, antibodies against voltage gated potassium channels have been suggested as a cause of acquired neuromyotonia in some patients.10 Autonomic dysfunction may be due to CNS immunoreactivity, as suspected in multiple system atrophy, or paraneoplastic syndrome.11 Disorientation, dementia, and psychosis in Morvan’s fibrillary chorea are similar to manifestations of limbic encephalitis. Symptomatic improvement with plasmapheresis, thymectomy, and chronic immunosuppression provides further support for an autoimmune or paraneoplastic basis.2 3
We present a patient with Morvan’s fibrillary chorea whose clinical presentation, and prolonged and sustained response to chronic immunosuppression, suggest an autoimmune aetiology or, furthermore, that Morvan’s fibrillary chorea is a paraneoplastic manifestation of cortical thymoma.
Case report
A 46 year old, right handed male machinist, with seasonal allergies and a 1 year history of recurrent atopic dermatitis, developed recurrent episodes of diffuse myalgias, abdominal pain, and diarrhoea. Five months later, he developed muscle cramps in lower limbs at rest, that worsened with exertion. Eight months later, the cramping spread to the trunk and upper limbs and was associated with weakness and fasciculations. The severity of the weakness progressed markedly over the next 2 years to the point where he required a cane to walk. Moderately severe dysphagia ensued. One year after the onset of weakness, he noted fluctuating left ptosis and diplopia. Four months later, he had recurrent exertion related chest pain and syncope associated with tachycardia. Excessive generalised sweating occurred 3 months later, followed within 2 months by severe constipation, urinary retention alternating with incontinence, confusional episodes with spatial and temporal disorientation, severe impairment of recent memory, agitation, severe anxiety, and insomnia. As his condition deteriorated, he developed increasing lower back pain, progressive leg weakness, persistent disorientation, inattention, and slurred speech. He lost 36 kg over 8 months, which was associated with decreased appetite. On admission to a hospital, he became more confused and agitated, and developed visual hallucinations. He was treated with meperidine, fentanyl, lorazepam, clorazepate, and haloperidol without improvement. Subsequently, he was transferred to our institution.
On physical examination, he had a blood pressure of 170/100 mm Hg, pulse rate of 118 beats per minute, respiratory rate of 27 breaths per minute, and a temperature of 38°C. Truncal hyperhidrosis was marked. He was awake, anxious, inattentive, disoriented to time and place, and extremely restless. Pupils were symmetric and reactive bilaterally. Left ptosis and mild bilateral lateral rectus and left medial rectus palsies were evident. He had continuous myokymia involving the face, trunk, and all limbs, with mild atrophy of the shoulder girdle and distal leg muscles. Moderately severe weakness of proximal and distal leg muscles was present with normal strength in the arms. Percussion myotonia was absent. Sensory testing and coordination were normal. Deep tendon reflexes were normal, except at the ankles where they were absent. Plantar reflexes were flexor. Atopic dermatitis involving the scalp, axilla, trunk, groin, and feet was present. The patient was intubated. Phenytoin was given and resulted in improvement of myokymia.
Laboratory testing disclosed a normocytic anaemia with a haemoglobin of 10 g/dl, packed cell volume of 30%, an erythrocyte sedimentation rate of 50 mm/h, a serum creatinine kinase concentration of 1245 u/l, with a normal relative index for the MB fractions, a lactate dehydrogenase concentration of 225 u/l (normal 90–200) with raised LD4 (12%) and LD5 (20%) fractions, and a serum ammonia of 87 μmol/l (normal 2–30). Serum electrolytes, calcium, magnesium, thyroid function, B12, rheumatoid factor, Lyme titre, lactic acid, pyruvic acid, erythrocyte transketolase, pseudocholinesterase, dilute Russell viper venom time, C3, C4, CH50, protein electrophoresis, immunoelectrophoresis, cryoglobulin, and antinuclear, anti-ds DNA, antismooth muscle, antineutrophil cytoplasmic, anticancer associated retinopathy, anti-Ri, antimyelin associated glycoprotein, antisulphatide, anti-GD1b, anti-GM1, anti-Yo, anti-Hu, anti-SS-A, anti-SS-B, ribosomal P protein, antithyroglobulin, antimicrosomal, antiphospholipid, and anticardiolipin antibodies were absent. A urine toxicology screen disclosed medications given before transfer. Cerebrospinal fluid examination showed a protein concentration of 44 g/dl, glucose of 82 mg/dl, 0 white blood cells/mm3, and 1 red blood cell/mm3. The CSF IgG index was raised at 0.9 (normal range <0.7). Oligoclonal bands and CSF antineuronal antibodies were absent. A CSF venereal research laboratory test was non-reactive. The chest radiograph was normal but chest CT with contrast showed an enlarged anterior mediastinal mass suspicious for thymic hyperplasia or thymoma. Computed tomography of the abdomen and pelvis and MRI of the lumbar spine were normal.
Confusion, anxiety, insomnia, and weakness persisted. Brain MRI and serial EEGs were normal. Electrocardiography and holter monitoring were normal. A two dimensional transthoracic ECG showed mild left atrial enlargement, and trace tricuspid and mild mitral regurgitation. An exercise treadmill test disclosed reversible inferior wall ischaemia. Cardiac catheterisation was normal.
Six days after admission to our hospital, plasmapheresis, using exchange volumes of 60 ml/kg/exchange, was done for five consecutive days. Left ptosis and extraocular muscle weakness resolved, and strength in the lower limbs improved. The patient received a second 5 day course of plasmapheresis. High dose oral prednisone at a daily dose of 1 mg/kg was started 16 days after admission. Trans-sternal thymectomy was performed 19 days after admission.
After thymectomy and on oral steroids, muscle strength continued to improve gradually, returning to normal 4 months after steroids were initiated. However, the patient had frequency of micturition. Urodynamic studies disclosed an areflexic bladder. He developed a recurrence of neuromyotonia and moderate weakness after steroids were tapered to 0.5 mg/kg/day. During this period, he developed psoriasis of the nails and arthritis of the hands and knees. Remission of weakness and arthritis was induced with an increase in steroid dosage. Azathioprine was started 3 months later (8 months after admission). Twenty four months after admission, all of his symptoms, including recent memory impairment, remain in remission on azathioprine and low dose oral steroids.
Methods
IMMUNOBLOTS
Serum striated muscle and anti-AChR antibodies (binding, modulating, and blocking) and N-type and P/Q-type calcium channel antibodies were measured by standard methodology (Mayo Clinic Laboratories). Serum antititin antibodies were tested in immunoblots using the MGT 30 peptide.12 Serum antiryanodine receptor antibodies were tested in immunoblots using myasthenia gravis serum diluted to 1:50 and peroxidase conjugated rabbit antibodies to human IgG diluted 1:500.13 Anti-voltage gated potassium channel antibodies were determined by immunoprecipitation of125I-α-dendrotoxin labelled extracts of human frontal cortex.10
PATHOLOGY
A biopsy of the anconeus muscle with intact motor nerve was obtained 5 days after admission, before plasmapheresis. Light microscopy examination was performed with the following stains: haematoxylin-eosin, PAS-haematoxylin, azure II methylene blue, adenosine triphosphatase, alkaline and acid phosphatase, and NAD-diaphorase, esterase, oil red O, myophosphorylase, and staphylococcal protein A-horseradish peroxidase conjugates for immune complexes. Electron microscopy of muscle was also performed. The thymus was examined under light microscopy.
Results
AUTOANTIBODIES AND IMMUNOBLOTS
Serum testing on admission detected binding anti-AChR antibodies at 7.5 nmol/l (normal range<0.4 nmol/l), modulating anti-AChR antibodies with 100% loss of AChR (normal range 0–20%), blocking anti-AChR antibodies with 27% blockade of AChR (normal range 0–25%), antistriated muscle antibodies at a titre of 1:960 (normal range<1:60), antititin antibodies at 1.4 optical density (OD) units (normal range 0 OD units), and anti-N-type calcium channel antibodies at 112 pmol/l (normal range<20 pmol/l). Anti-P/Q-type calcium channel and antiryanodine receptor antibodies were undetectable. Serum anti-voltage gated potassium channel antibodies were raised (manuscript in preparation).
ELECTROPHYSIOLOGICAL STUDIES
Nerve conduction studies and EMG were performed 1 day after admission and disclosed the following: (1) normal motor and sensory nerve conduction studies, (2) multiple repetitive compound motor action potentials to single stimulation, (3) no decremental response to 2 Hz stimulation of the left axillary nerve, (4) doublets, triplets, multiplets, and continuous neuromyotonic discharges of a frequency close to 200 Hz (fig 1A, B) in multiple muscles without evidence of denervation, (5) a 42% increment in the area of the compound muscle action potential in response to 30 Hz stimulation of the left ulnar nerve (fig 1D), and (6) poor identification of the F wave and H reflex due to continuous muscle activity and obliteration of the silent period. Nerve block of the right common peroneal nerve with 1% xylocaine failed to eliminate neuromyotonia in the right tibialis anterior (fig 1C). Single fibre EMG was not performed due to agitation. A repeat EMG was performed 3 weeks after the initial study, after plasmapheresis and thymectomy. Repetitive stimulation of the facial nerve, with recording from the nasolabialis, and of the circumflex nerve, with recording from the mid-deltoid, showed no electrodecremental response. In vitro microelectrode studies were performed on biopsied anconeus muscle fibres. A reduction in the amplitude and frequency of spontaneous miniature end plate potentials and a marked reduction in mean end plate potential quantal content was demonstrated.14

Electrodiagnostic findings. (A) Nerve evoked neuromyotonia was recorded with surface electrodes over the abductor hallucis muscle in response to electrical stimulation of the right posterior tibial nerve. Calibration: 500 μV dot to dot vertically and 10 ms dot to dot horizontally. (B) A spontaneous run of neuromyotonic discharges with an intraburst frequency of 160 Hz was recorded with a monopolar needle electrode in the right medial gastrocnemius muscle. Calibration: 200 μV dot to dot vertically and 10 ms dot to dot horizontally. (C) A spontaneous burst of repetitive motor unit potentials with an intraburst frequency of 100 Hz was recorded from a needle electrode in the tibialis anterior muscle. This discharge was resistant to nerve block. Calibration: 500 μV dot to dot vertically and 10 ms dot to dot horizontally. (D) 30 Hz repetitive stimulation of the right ulnar nerve resulted in a 42% increment in the area of the compound muscle action potential. Calibration: 2 mV dot to dot vertically and 50 ms dot to dot horizontally.
PATHOLOGY
Frozen sections of transversely oriented anconeus muscle disclosed evidence of both denervation and fibre necrosis with regeneration. Denervation changes included scattered angular and atrophic mixed type 1 and 2 fibres arranged individually or in small groups, with associated clumps of pyknotic sarcolemmal nuclei and focal proliferation of endomysial collagen (fig 2). No fibre-type grouping or target fibres were seen. Groups of up to 20 pale necrotic or basophilic regenerating fibres were scattered randomly throughout the specimen and were associated with numerous macrophages that totally engulfed many fibres (fig 3). Electron microscopy confirmed the necrosis of muscle fibres and absence of abnormalities in peripheral nerves included in the specimen.
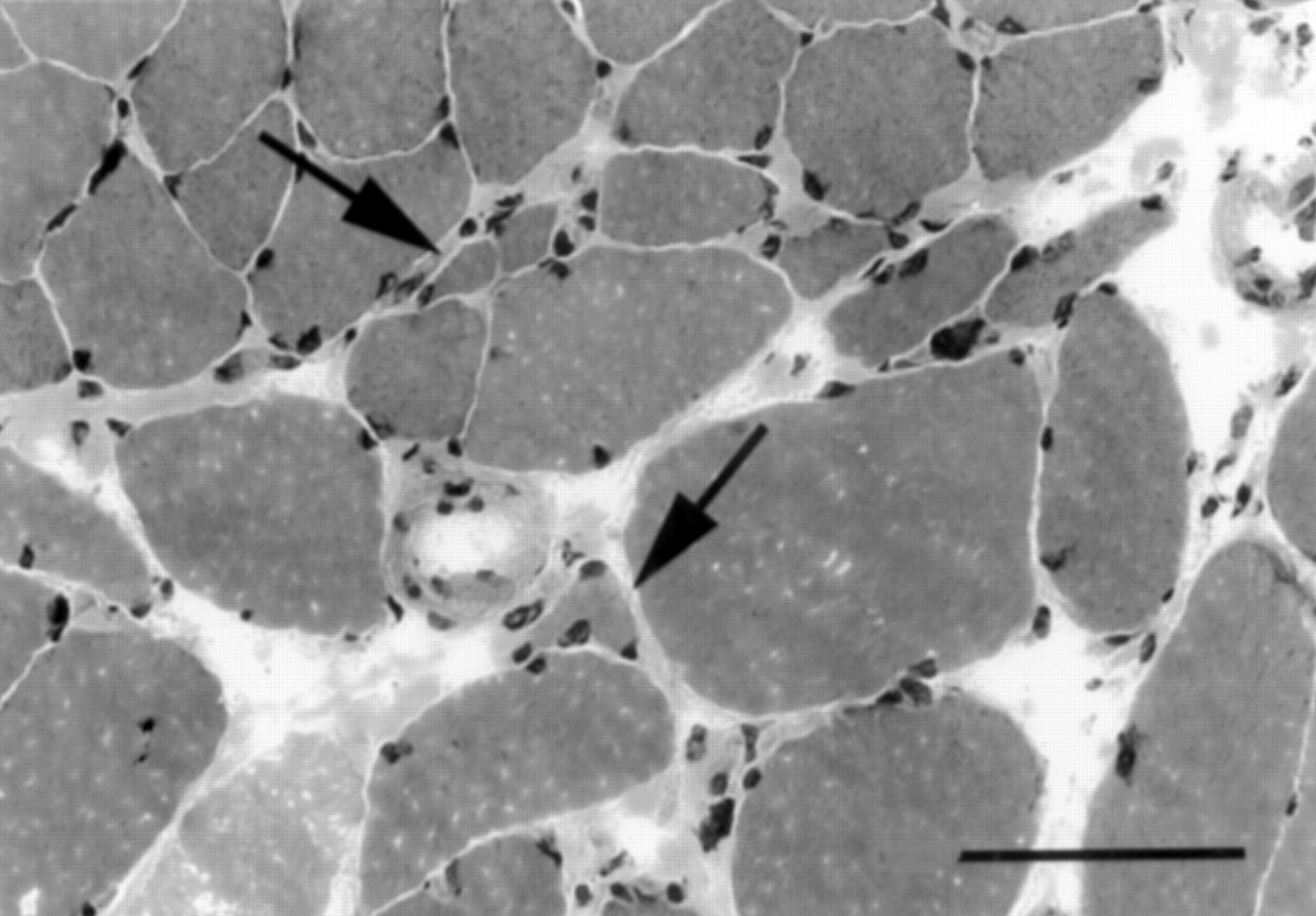
Transverse section of biopsied anconeus muscle shows evidence of denervation that includes multiple angular atrophic fibres lying in small groups or singly (arrows), increase in endomysial collagen around the atrophic fibres, and scattered clumps of small dense sarcolemmal nuclei. (Haematoxylin-eosin stain. Original magnification×200; bar=100 μm.)

{kind=link}
{kind=link}
{kind=link}
Transverse section of biopsied anconeus muscle shows randomly distributed foci of necrotic muscle fibres and macrophage accumulations (arrows), as well as numerous foci of basophilic regenerating fibres. (Haematoxylin-eosin stain; original magnification×250; bar=50 μm.)
Light microscopic examination of the thymus disclosed benign, non-invasive cortical thymoma.
Discussion
The patient described above exhibited the cardinal manifestations of Morvan’s fibrillary chorea: (1) neuromyotonia, which occurred early during the course of illness and was characterised by muscle cramping and myokymia; (2) dysautonomia presenting as cardiac syncope, severe constipation, and urinary overflow incontinence; (3) hyperhidrosis of the trunk and limbs; (4) anorexia, resulting in an 36 kg weight loss over 8 months; (5) encephalopathy, which developed later in the course of illness and was characterised by spatial and temporal disorientation, impairment of recent memory, anxiety, and restlessness; (6) severe insomnia; (7) visual hallucinations; and (8) pruritis with atopic dermatitis. These symptoms indicate involvement of central, autonomic, and peripheral nervous systems. In addition, our patient had defects in both presynaptic and postsynaptic neurotransmission by in vitro electrophysiology, which correlated with the presence of anti-N-type voltage gated calcium channel and anti-AChR antibodies respectively. Myopathy, as indicated by muscle histopathology and raised creatinine kinase concentrations, correlated with detectable serum antibodies to cytoskeletal and cytoplasmic muscle proteins, including rapsyn, syntrophin, and proteins with molecular weights similar to myosin (200 kDa), sarcoplasmic and endoplasmic reticulum calcium ATPase (110 kDa), triadin (95 kDa), calsequestrin (63 kDa), and troponin T (38 kDa).15 Peripheral neuropathy, which was suggested by the symmetric absence of ankle reflexes, was more definitively indicated by denervation changes on muscle histopathology. Psoriasis of the nails, a neuroimmunocutaneous disorder, was an associated finding.
To date, only four cases of Morvan’s fibrillary chorea have been reported in the English literature. Morvan originally described five patients with the disease. They had continuous fibrillary contractions of the limb muscles, muscle cramping, fatigue, and varying degrees of hyperhidrosis, anorexia, insomnia, pruritis, tachycardia, tachypnoea, diarrhoea and psychosis.1 Fischer-Perroudon et al reported a patient with Morvan’s fibrillary chorea who had absolute insomnia for 4 months, and nocturnal visual and auditory hallucinations that improved after insomnia was treated with massive doses of 5-hydroxytryptophan.9 Halbach et alreported two patients with thymoma and anti-AChR antibodies, one of whom also had a raised carcinogenic embryonic antigen concentration.2 Madrid et al described a patient with prostatic adenoma and in situ carcinoma of the colon who had evidence of peripheral neuropathy only by electrophysiology and muscle histopathology.3 Although symptoms may remit spontaneously, Morvan’s fibrillary chorea is a potentially fatal disorder, with the cause of death being unclear in most cases.1 2 9
Myasthenia gravis is one of the best characterised autoimmune disorders which can develop as a paraneoplastic syndrome.16 As in myasthenia gravis, a paraneoplastic aetiology for Morvan’s fibrillary chorea in our patient is inferred by several findings: (1) profound neurological disability preceding the diagnosis of thymoma, (2) the presence of thymoma with high titres of anti-AChR, antistriated muscle, antititin, antivoltage gated potassium channel antibodies, and anti-N-type calcium channel antibodies, (3) multiple coexistent autoimmune disorders, and (4) a dramatic response to plasmapheresis, thymectomy, and chronic immunosuppression. Tumours such as thymoma, prostatic adenoma and in situ carcinoma of the sigmoid colon have been found in patients with Morvan’s fibrillary chorea when a paraneoplastic aetiology has been suspected.2 3Autoantibodies have been detected in patients with Morvan’s fibrillary chorea, including anti-AChR antibodies in patients with thymoma and a raised CSF IgG index and oligoclonal bands in patients with severe insomnia and hallucinations.2 3 9 In our patient, it is unlikely that hepatic dysfunction caused encephalopathy as the EEG was normal and symptoms persisted after correction of the serum ammonia. We suspect that there may be other, as yet undetected, autoantibodies in patients with Morvan’s fibrillary chorea.
The neurological components of the disease may be attributed to antibody mediated immune responses to various target antigens. Neuromyotonia, one of the earliest and best studied manifestations of Morvan’s fibrillary chorea, is characterised by muscle twitching and cramping at rest, with exacerbation on exercise. It has a distinct electrophysiological pattern which is characterised by repetitive motor unit potentials firing in rhythmic bursts. The origin of neuromyotonia is not clearly established. Isaacs implicated the peripheral nerve terminal when he demonstrated persistence of continuous muscle activity during general anaesthesia and after proximal nerve block and termination with curare blockade of neuromuscular transmission.17 Subsequent studies investigating the effect of nerve block on neuromyotonia have disclosed variable results. In some patients, nerve block resulted in abolishment, partial reduction, or no effect, suggesting that neuromyotonic discharges may be generated from various sites along the length of the nerve.2 4 17-26 Failure to abolish neuromyotonia with nerve block in our patient suggests that neuromyotonia is arising from distal nerve terminals, or even the muscle itself. Clinical and electrophysiological evidence of peripheral neuropathy may be absent. Our patient had normal nerve conduction studies although muscle histopathology disclosed changes indicative of chronic denervation. Neuromyotonia has been associated with various peripheral neuropathies, including axonal neuropathy, radiation induced neuropathy, hereditary motor neuropathy, and the neuronal type of Charcot-Marie-Tooth disease.17 26 28-31 It has also been associated with autoimmune and paraneoplastic diseases, including Guillain-Barré syndrome, myasthenia gravis, thymoma, lung cancer, plasmacytoma with IgM paraproteinaemia, primary amyloidosis with IgG monoclonal kappa spike, heavy metal intoxication, and anti-AChR, antithyroid, anti-GM1, antinuclear, and anti-DNA antibodies.4 6 10 26-30 32 34-36 Antibodies directed against voltage gated potassium channels have been reported in some patients with acquired neuromyotonia, and the node of Ranvier has been proposed as a possible target.10 The relation between anti-voltage gated potassium channel antibodies and thymoma is not clear, although some cases of neuromyotonia have been associated with thymoma.30 Other manifestations of Morvan’s fibrillary chorea may also have a paraneoplastic or autoimmune basis. Myopathy and peripheral neuropathy have been associated with paraneoplastic and autoimmune diseases. Extraocular, bulbar, and limb muscle weakness, may result from paraneoplastic dysfunction of presynaptic or postsynaptic neuromuscular transmission. Raised serum titres of anti-N-type calcium channel antibodies in our patient support the hypothesis that Morvan’s fibrillary chorea is a paraneoplastic syndrome. Anti-N-type calcium channel antibodies are found in 49% of patients with Lambert-Eaton syndrome, and in 54% of patients with a paraneoplastic encephalomyeloneuropathy complicating lung, ovarian, or breast carcinoma, albeit in lower titres.37 Most patients with Lambert-Eaton myasthenic syndrome have anti-P/Q-type calcium channel antibodies whereas anti-N-type calcium channel antibodies have been associated with malignancy, usually in the presence of anti-P/Q-type calcium channel antibodies.37 Acquired dysautonomia may also be a manifestation of paraneoplastic syndrome, as release of acetylcholine from parasympathetic neurons is predominantly coupled to calcium influx through N-type calcium channels.38 39Alternatively, acquired dysautonomia and severe insomnia may be associated with antibodies directed against a specific CNS site, as reported in multiple system atrophy.11 Limbic encephalitis, which is characterised by encephalopathy and psychosis, may be a component of Morvan’s fibrillary chorea. The presence of a raised CSF IgG index and oligoclonal bands in patients with Morvan’s fibrillary chorea with severe insomnia and hallucinations suggests immune mediated limbic dysfunction.
Cortical thymomas are associated with various neurological autoimmune diseases, most commonly myasthenia gravis. Some patients with Morvan’s fibrillary chorea who had anti-AChR antibodies, but no clinical manifestations of myasthenia gravis, were found to have thymoma. Giant cell myositis, myocarditis, and intractable ventricular tachycardia have been reported in patients with benign and malignant thymoma.40-42 Antititin antibodies, which are mainly directed against a myofibrillar protein in striated muscle, is highly specific for myasthenia gravis with thymoma and associated with more severe disease in patients with late onset myasthenia gravis.43 44 These antibodies correlate with the myopathic changes evident on muscle histopathology. Antititin antibodies may also conceivably cross react with epitopes on the heart and induce myocarditis and ventricular arrhythmias. Other conditions associated with malignant thymoma include acquired dysautonomia and limbic encephalitis.45-48 Some patients with limbic encephalitis have peripheral and central autoantibodies that react to human and rat brain, and newborn rat thymus.47 48
The potentially fatal outcome of Morvan’s fibrillary chorea underscores the importance of early diagnosis and treatment, as well as an aggressive search for thymoma or neoplasm. Experience in the treatment of Morvan’s fibrillary chorea has been limited. Chronic immunosuppression has resulted in marked symptomatic improvement in one patient, whereas transient relief has been reported in two patients after plasmapheresis.2 3 Acutely, plasmapheresis improved neuromyotonia and muscle strength in our patient. Sustained remission was induced only after thymectomy and long term immunosuppression with prednisone and azathioprine. Our experience suggests that immunosuppressive therapy plays an important part in the management of patients with Morvan’s fibrillary chorea. Dramatic and prolonged remission with these treatments supports our hypothesis that Morvan’s fibrillary chorea is a paraneoplastic manifestation of thymoma.
In summary, our patient is the first reported case of Morvan’s fibrillary chorea in whom the following concomitant conditions are present: (1) clinical manifestations of myasthenia gravis, in addition to anti-AChR antibodies and thymoma, (2) presynaptic electrophysiological defects of neuromuscular transmission and autoantibodies directed against the presynaptic junction, (3) myopathy, associated with autoantibodies to muscle, and (4) psoriasis and atopic dermatitis. A dramatic improvement in symptoms was achieved acutely with plasmapheresis, whereas prolonged and sustained remission was induced with thymectomy and chronic immunosuppression. Collectively, our patient’s presentation and laboratory findings support the following conclusions: (1) Morvan’s fibrillary chorea falls into the range of neurological manifestations of cortical thymoma, (2) Morvan’s fibrillary chorea is most likely a paraneoplastic complication of cortical thymoma, and (3) treatments traditionally employed for paraneoplastic autoimmune disorders induce dramatic remission of the disease.