Article Text

Abstract
This paper describes a 20 year old woman with a new combination of neurological impairments in which the motor phenomena were responsive to corticosteroid treatment. She had lifelong moderate learning impairment. A variable ataxia with cerebellar characteristics was present from early life, with early severe exacerbation when seizures were uncontrolled. Atypical absence and simple and complex partial seizures were present from the first year of life and EEG abnormalities were maximal in the right parietal region, concordant with a mild non-specific abnormality of the white matter in the region of the trigone. Episodes of alternating hemiplegia occurred from 11 years, unassociated with seizures. Exercise induced dystonia occurred from the age of 5. After 10–20 minutes walking, her right foot would turn in and the right leg would stiffen, followed by the left and by falling and inability to get up for several minutes. Prednisolone improved her ataxia and was associated with cessation of both seizures and exercise induced dystonia. This adds a new syndrome to the corticosteroid responsive motor disorders associated with epilepsy.
- exercise induced dystonia
- steroid dependent
- epilepsy
- alternating hemiplegia
Statistics from Altmetric.com
The disabilities that coexist with epilepsy have often been regarded as direct effects of brain damage. However, selective and global cognitive deficits without evidence of cerebral lesions on high quality MRI occurring in the Landau-Kleffner syndrome1 2indicate the presence of other dynamic mechanisms, which seem to inactivate cerebral systems through functional rather than structural mechanisms. This concept has been extended from language functions to include motor organisation or praxis involving gait.3 We suspect that some motor impairments that occur with epilepsy are as yet not fully described, nor are the mechanisms defined. For example, dystonic phenomena were seen in the two children described with loss of praxis.3 We describe a new dystonic phenomenon in a young woman who had a range of cognitive and motor disorders in association with epilepsy.
Case history
This 20 year old woman was the third child of a family with no history of epilepsy. Her mother and maternal aunt had classic migraine but without hemiplegic symptoms. The pregnancy was accompanied by mild “influenza” at two months (the rest of the pregnancy and birth were normal with a birth weight of 3.2 kg). Her history will be described under problem headings.
LEARNING IMPAIRMENT
She was described as being bright and normal until the last part of the first year, when seizures began. Her subsequent cognitive progress was slow but she had never regressed cognitively. She was able to put words together from 1.6 years but speech was slow and indistinct for a further 2 years. Psychological assessment at 18 showed IQ scores on the Wechsler scale (WAIS) of verbal 71, performance 54, and full scale 62. Her educational progress was consistent with moderate learning disability.
ATAXIA
She first walked independently, although unsteadily, at 1.8 years. She had walked round the furniture from 8 months but that skill and her crawling, sitting, and other purposeful movements had been temporarily lost with the onset of seizures. For several months her global physical abilities regressed markedly. Her parents used reins to assist walking until 4 years. Repeated examinations disclosed an intention tremor and broad based gait which showed considerable variability and was regarded as cerebellar. Physical signs included slurred speech with slowness of alternating movements of the tongue. The upper limbs showed an intention tremor. There was no sensory loss. She had a broad based gait but was stable in the household situation. Outdoors her walking was accompanied by falls or “collapses”. In the lower limbs there was normal tone, with brisk tendon reflexes and flexor plantar responses. These signs have not changed over the years.
EPILEPSY
At 8 months when trying to crawl she fell on to her nose. Her eyes looked vacant and rolled. The episode lasted for 10 minutes. Her mother thought that subsequent episodes could be aborted by a smack or a pinch and she would laugh if tickled in an attack. The early episodes were particularly related to intercurrent infection. Some consisted of flicking of the eyes up and to one side (“darting about”) for a few seconds, accompanied by a fall and often a scream. An EEG at this time was reported as normal. Sodium valproate, given at 1.6 years, stopped the attacks. She became brighter and steadier on her feet and walked independently by 1.8 years. Complex absences returned at 7 years when sodium valproate was withdrawn because of anorexia. Her school progress slowed at that stage. Ethosuximide relieved seizures but did not improve her development. At 8 years a single new type of “seizure” occurred in which her left arm went into a “cramp-like” position with flexion at the elbow and extension at the wrist and her legs gave way. Consciousness was retained. This was followed by transient left sided weakness. The next year absences recurred at a high rate; sodium valproate was reintroduced, but the seizure rate was not reduced by this or carbamazepine and clobazam. School progress stopped and her speech was more slurred. However, when ethosuximide and sodium valproate were used in combination, better, but not complete, seizure control was achieved with improvement in speech and school performance. Her family also reported involuntary flinging movements of the left arm when dropping off to sleep.
EPISODES OF HEMIPLEGIA
At 11 years, she had the first attack of abrupt onset of paraesthesiae of the left hand and foot with left facial weakness, slurred speech, and some weakness of the left arm and leg lasting for 20 minutes. These episodes tended to be followed by vomiting and sleep. The episode at 8 years (reported under “epilepsy”) had been accompanied by abnormal postures in the left arm and were categorised as either focal seizures with Todd’s paresis or hemiplegic migraine.
At 13 years she had episodes of dizziness and inability to walk lasting 45 minutes without disturbance of consciousness. In a later single attack, this was accompanied by weakness of the right arm and great distress. One episode of left hemiplegia followed a trivial argument in which she cried and on two occasions the possibility that hyperventilation was contributing to the clinical picture was considered. On another occasion it was clear that a breath holding attack after being “told off” was the trigger for an episode of hemiplegia. At the end of these episodes she was very confused, vomited violently, and took several hours to recover. Although the episodes of hemiplegia and ataxia were not immediately related to clinical seizures, they occurred when seizures were poorly controlled. A restricted diet failed to produce sustained benefit. Further right and left sided hemiplegic attacks occurred. Each of these lasted for several hours; they were followed by pallor and vomiting. She had generally very poor fine and gross motor organisational skills throughout this time.
In summary, she had about 10 episodes of dense flaccid hemiparesis, mostly on the right, from the age of 9 years lasting about one hour, mostly in the afternoon, associated with inability to speak and followed by confused speech. They were often associated with flashing lights and occasionally associated with initial dysaesthesia and usually followed by vomiting and contralateral headaches. No jerking has been noticed in these episodes. Recovery was complete after each episode.
EXERCISE INDUCED DYSTONIA (MOTOR EPISODES DURING WALKING)
These were noticed from the age of 5. Their description by history and video was consistent. From 5 years when she could walk a reasonable distance she would, after 10–20 minutes, tend to hold someone’s hand, her right foot would turn in and she would stiffen below the knee, the left leg would follow into a similar posture and she would eventually fall and be unable to rise for several minutes. Unless she rested for at least 30 minutes, the dystonic posture would rapidly return and spread to the other leg. On one occasion this lasted for 3 hours. There was no loss of consciousness and the frequency was totally dependent on the amount of extended walking that she did. These collapses occurred on most days unless she restricted her walking distance. They often occurred several times daily.
BEHAVIOUR
Throughout childhood she was a friendly, cooperative girl without notable behaviour problems.
EEG FINDINGS
EEGs at various stages showed the following:
2.5 years—Excess of slow components with non-localised sharp elements.
5.5 years—Infrequent paroxysmal features, occasionally forming spike-wave complexes with a mild degree of photosensitivity.
5.10 years—A single burst of generalised 3.5 second polyspike and slow waves.
8.7 years—Multifocal discharges over the posterior temporal and parietal regions independently on the right and left.
10.1 years—Isolated mid-parietal discharges spreading over the two hemispheres with, in addition, some prolonged runs of 3/s spike-wave complexes.
15.7 years—No definite abnormality.
19 years—EEG performed during episodes of exercise-induced dystonia showed no discharges associated with the event.
19.4 years—Ill formed sharp waves as bursts over both hemispheres.
There was a right parietal emphasis to these abnormalities.
Four sleep recordings were made at ages 18.1 years, 18.3 years, 18.4 years, and 21.5 years and did not show evidence of electrical status epilepticus.
INVESTIGATIONS
Brain MRI at 15 and 19 years of age showed mild non-specific high signal in the white matter at the right trigone (figure). The following were normal : MR angiography, spine MRI, transcranial Doppler of basal arteries, eye movement monitoring, electroretinography and visual evoked potentials, cardiac echocardiography, vacuolated lymphocytes, electrolytes, calcium, alkaline phosphatase, ammonia, lactate, very long chain fatty acids, ceruloplasmin, copper, transferrin electrophoresis, immunoglobulins, α-fetoprotein, biotinidase, autoimmune profile, blood amino acids, urinary organic acids, chromosomes including fragile X, DNA for MELAS mutation, and CSF lactate.

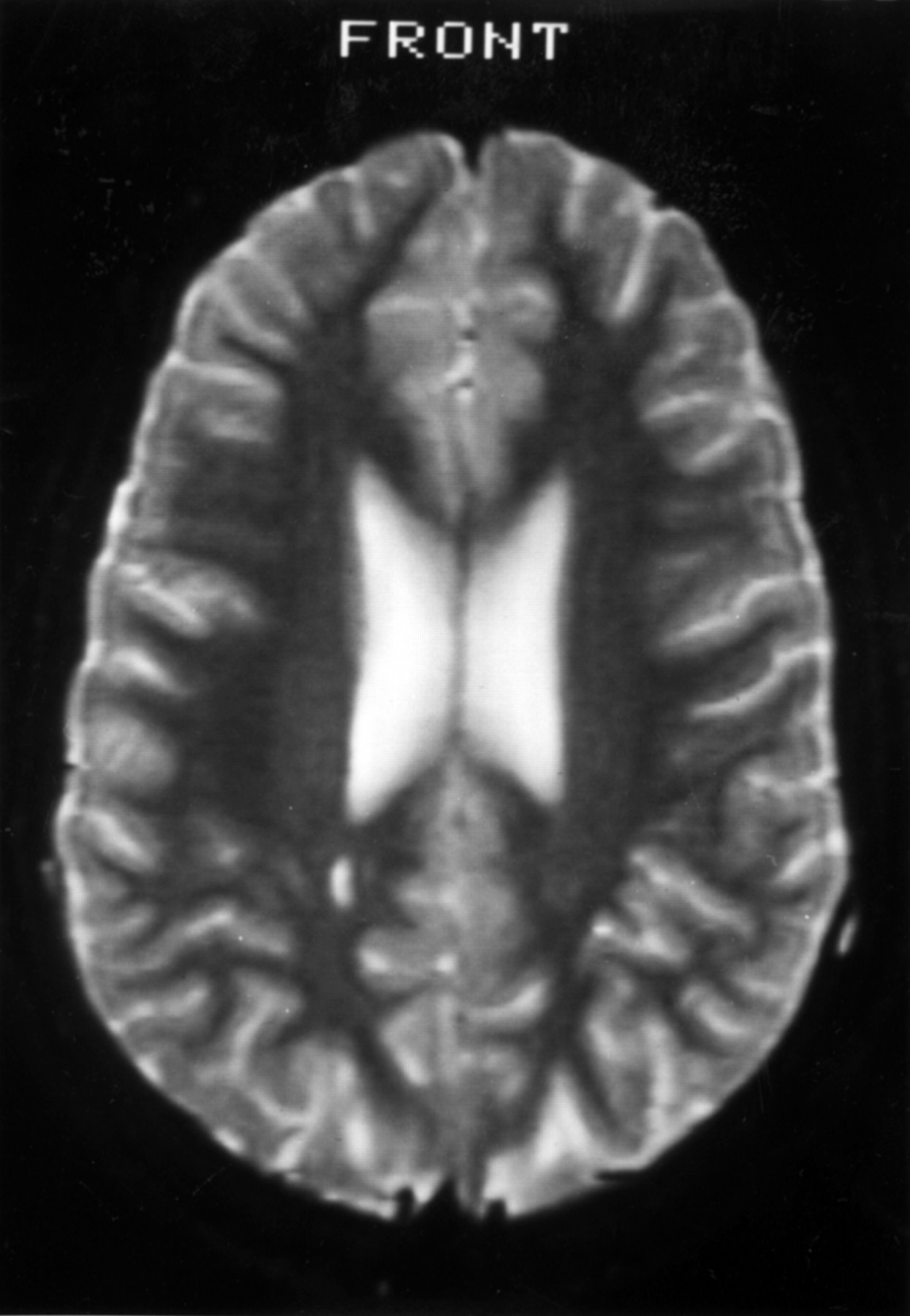{kind=link}
MRI showing mild abnormality of the white matter in the region of the right trigone.
TREATMENT WITH CORTICOSTEROIDS
At the age of 20 she had a short course of prednisolone (1.5 mg/kg for 10 days) followed by 25 days on a reducing dose. During this time she was free of falls and her walking improved, but the falls returned after the treatment was stopped. A second similar course was given. After 2 days she was walking better and prolonged walking produced no falls; the seizures stopped, her speech was clearer, the intention tremor resolved, and she was generally brighter but relapsed on withdrawal.
Discussion
This patient with a minor right parietal abnormality on MRI developed epilepsy with both focal and generalised seizures with frequent atypical absences from towards the end of the first year of life. The EEG showed multifocal abnormalities. Her most notable disability was moderate global cognitive delay. The motor abnormalities included a long term variable ataxia which had the appearance of a cerebellar deficit, loss of fine motor function, dystonic lower limb episodes with “collapses” on walking from the time of being able to walk long distances, and episodes of hemiplegia with symptoms suggesting a vascular/migrainous mechanism. The attack at 8 years provides the most interesting linkage of the various phenomena : the left arm went into a dystonic posture, the legs gave way and there was a transient postictal left hemiparesis. However, none of the phenomena of the characteristic absence seizures occurred in that attack. The dystonic phenomena on walking long distances resemble those of the rare disorder of paroxsysmal exercise induced dystonia or paroxsysmal dystonic choreoathetosis, although the collapse aspect is atypical and this is not a disorder associated with epilepsy.4 5
The EEGs showed both generalised and focal interictal discharges with an emphasis in the right parietal region close to the area of white matter abnormality seen on MRI. It is important to note that there was no seizure activity during the exercise induced dystonia.
Early treatment with sodium valproate produced amelioration of the seizures and pervasive symptoms. Corticosteroids were used because of their dramatic effect on the two previous children reported with severe generalised apraxia associated with epilepsy.3 One of these children, who had acquired aphasia typical of the Landau-Kleffner syndrome, had a single episode of holding one arm above her head in an odd posture for 3 days. The second child could not initially walk because of the appearance of dystonic postures and jerks in the lower limbs. In children, corticosteroids are used for severe epilepsies more commonly than in adults. The effect of corticosteroids in this case was dramatic on seizures, ataxia, motor organisational problems, and exercise induced dystonia and collapses. These collapses showed a stereotyped sequence of progressive dystonic posturing of the feet with the legs eventually giving way, causing falling, after which the legs would not work for 30 minutes or more. No hypothesis is offered for why a seizure related phenomenon should be exercise induced, although a cerebral perfusion steal or inappropriate hyperventilation are possible. However, a possible mechanism for the episodes of hemiplegia can be offered. In Landau-Kleffner syndrome, temporal hypoperfusion is commonly seen using single photon emission computerised tomography (SPECT)6 and hypometabolism using PET. It is possible that some types of epilepsy have major effects through selective paroxysmal hypoperfusion and that it is immaterial whether a triggering seizure is found. In our experience of a large series of children with Landau-Kleffner-like syndromes, Todd’s pareses, and other transient motor phenomena are common. It may well be that there is a group of seizure disorders in which the internal cerebral neurovascular effects are more important than the traditional clinical paroxysmal seizures. Our reasons for keeping them within the general rubric of epilepsy are the strong association with clinical and electrophysiological seizures, the continuum of such motor events in people with epilepsy, and the ready reversibility of the phenomena on treatment opens the possibility of benefit of antiepileptic treatments including corticosteroids. Corticosteroids may prove a useful diagnostic test but an unacceptable treatment in some cases.
The possibility exists that a genetically determined channelopathy or cerebral vascularopathy could explain some of the phenomena, but we have no supportive evidence of either. Specifically, there was no skin or other system involvement to suggest Sneddon’s syndrome.7