Article Text

Abstract
OBJECTIVES To assess the extent of loss of myelinated nerve fibres and spinal motor neuron loss in chronic inflammatory demyelinating polyneuropathy (CIDP), a clinicopathological study was conducted on biopsied sural nerves and necropsied spinal cords from patients with CIDP.
METHODS The myelinated fibre pathology of 71 biopsied sural nerves and motor neuron pathology of nine necropsied spinal cords at L4 levels in patients with CIDP were quantitatively and immunohistochemically assessed.
RESULTS Myelinated nerve fibre density was significantly diminished to 65.4% of the control values (p <0.0001), correlating inversely with the extent of segmental demyelination and remyelination (r = −0.43, p < 0.0005) and duration of illness (r = −0.31, p < 0.01). Numbers of large spinal motor neurons in CIDP were variably but significantly diminished (range from 46.0 to 97.6% of the age matched control value (p < 0.005)), and reactive astrogliosis was evident in the ventral horn in CIDP. The frequency of ventral horn neurons exhibiting central chromatolysis and the accumulation of phosphorylated high molecular weight neurofilament protein was significantly higher in CIDP than in controls (p<0.01 and p<0.05).
CONCLUSIONS The loss of nerve axons and spinal motor neurons is common in CIDP, and extensive in some cases. These neuronal and axonal losses may influence the functional prognosis in CIDP.
- chronic inflammatory demyelinating polyneuropathy
- axon loss
- spinal motor neuron
Statistics from Altmetric.com
The pathological hallmark of chronic inflammatory demyelinating polyneuropathy (CIDP) is segmental demyelination and mononuclear inflammatory cell infiltration in the peripheral nerves, accompanied by varying degrees of axonal degeneration, myelinated fibre loss, and endoneurial oedema.1 Regarding the pathology of the CNS in CIDP, central chromatolysis in spinal motor neurons has often been reported,2-6 and sporadic cases with a slight loss of spinal motor neurons have occasionally been documented.3 6-9 Spinal motor neuron loss as well as nerve myelinated fibre loss could be important factors influencing functional recovery. In this study, we assessed the degree of involvement of spinal motor neurons and peripheral nerve axons in CIDP.
Methods
SPECIMENS
After informed consent was given, sural nerve biopsy specimens from 71 patients with CIDP (50 males and 21 females) were obtained at the Nagoya University School of Medicine and its affiliated hospitals over 11 years. Age at biopsy ranged from 2 to 81 years; mean (SD) age 48.5 (21.9) years. The duration of illness before biopsy ranged from 2 months to 28 years; mean (SD) 2.9 (5.8) years. The spinal cords were obtained at necropsy from nine patients with CIDP. Three of these patients were necropsied at the Nagoya University Hospital and affiliated hospitals, and others were necropsied in hospitals located throughout Japan during the past 11 years. These patients consisted of six men and three women, aged 49 to 73 years; mean (SD) 62.4 (8.9) years. Their duration of illness ranged from 4 months to 8 years. Clinical profiles of necropsy cases are summarised in table1.10
Clinical features of necropsied patients with CIDP
The diagnosis of CIDP in our study was assessed using the criteria of Barohn et al 11 or the ad hoc subcommittee of the American Academy of Neurology.12
ASSESSMENT OF SURAL NERVE BIOPSY
Sural nerve biopsy specimens were fixed in glutaraldehyde in 0.025 M cacodylate buffer (pH 7.4) and embedded in epoxy resin. Semi-thin sections were stained with toluidine blue, and the density of myelinated fibres was analysed quantitatively using a computer assisted imaging system (Luzex FS, Nireco, Tokyo, Japan). The extent of subperineurial oedema was assessed as an increase in the subperineurial space by comparing the subperineurial area to the total endoneurial area using the same imaging system. A part of the nerve specimen was processed for teased fibre analysis, and the condition of each fibre was assessed according to our previously indicated criteria.13 A portion of the nerve specimen was fixed in 10% buffered formalin and embedded in paraffin, and then processed for immunohistochemical study. Mouse monoclonal antibodies (mAb) to human leucocyte common antigen (LCA, DAKO, Denmark; dilution, 1:50), helper/inducer T cells (CD4, Novocastra, UK; dilution, 1:10), cytotoxic/suppressor T cells (CD8, DAKO; dilution, 1:25), and macrophages (CD68, DAKO; dilution, 1:10) were used. The avidin-biotin-peroxidase complex (ABC) method was performed using a Vectastain Kit (Vector Laboratories, Burlingame, CA, USA) as described previously.14 The extent of endoneurial and epineurial inflammatory infiltrates was assessed in a blind fashion for each mAbs.
Eleven sural nerve specimens from subjects with non-neurological diseases (eight men and three women, aged 28 to 71 years; mean (SD) 44.6 (13.5) years) obtained at biopsy for transplantation or at necropsy performed within 3 hours of death were processed in the same way as the biopsy specimens from patients with CIDP, and served as controls.
ASSESSMENT OF SPINAL CORD PATHOLOGY
The spinal cord was removed at necropsy, fixed in 10% buffered formalin solution, and processed for paraffin sections. Routine histological studies with haematoxylin and eosin, Klüver-Barrera, and Bodian stain were performed on lumbar spinal cord specimens. The population of spinal motor neurons was assessed as described previously.15 16 From the rostral end of the fourth lumbar segment, up to 300 serial 10 μm thick sections were prepared, and every 10th section was stained with the Klüver-Barrera technique. Photomicrographs were taken at a magnification of ×205, including the left ventral horn in each stained section. The spinal ventral horn was defined as the area of grey matter ventral to a line through the central canal perpendicular to the ventral sulcus. Diameters of all neurons with obvious nucleoli were measured on photomicrographs using a particle size analyser (TGZ-3, Carl-Zeiss, Germany). The neurons were classified arbitrarily into three groups according to their diameters as large (⩾32.8 μm), medium sized (⩾24.8 μm to <32.8 μm), and small (<24.8 μm) neurons.15 16 Neuronal numbers were expressed as the total number of cells/50 sections as described previously.15 16 Immunohistochemical assessment was performed in the same manner as for the sural nerves. We used a mAb Ta-51 specific to a phosphorylated epitope of high molecular weight subunits of neurofilaments (pNFH; dilution, 1:10), a mAb against human glial fibrillary acidic protein (GFAP, DAKO; dilution, 1:200), human B cells (CD20, DAKO; dilution, 1:50), and a broad range of T cells (UCHL1, DAKO; dilution, 1:125). CD68, CD4, CD8, and LCA antibodies were used in the same manner as the sural nerves. To assess the occurrence of motor neurons expressing pNFH in neuronal perikarya, Ta-51 positive ventral horn neurons with obvious nucleoli were counted, and a ratio to the total neuronal population was expressed as described previously.14 17 Gliosis in the ventral horn was assessed using Holzer stain and immunohistochemistry for GFAP. Motor neurons satisfying the criteria for central chromatolysis by Campbell and Novick18 were designated as undergoing active central chromatolysis, and their occurrence among total neurons was estimated on Klüver-Barrera stained sections as described before.15
Spinal cords of seven subjects who died of non-neurological disorders, (aged 47 to 81 years; mean (SD) 66.6 (12.7) years), were examined in the same manner as age matched controls.
STATISTICAL ANALYSIS
Statistical analyses were by Student’st test, Mann-WhitneyU test, and Pearson’s correlation coefficient; p<0.05 was taken as significant.
Results
SURAL NERVES
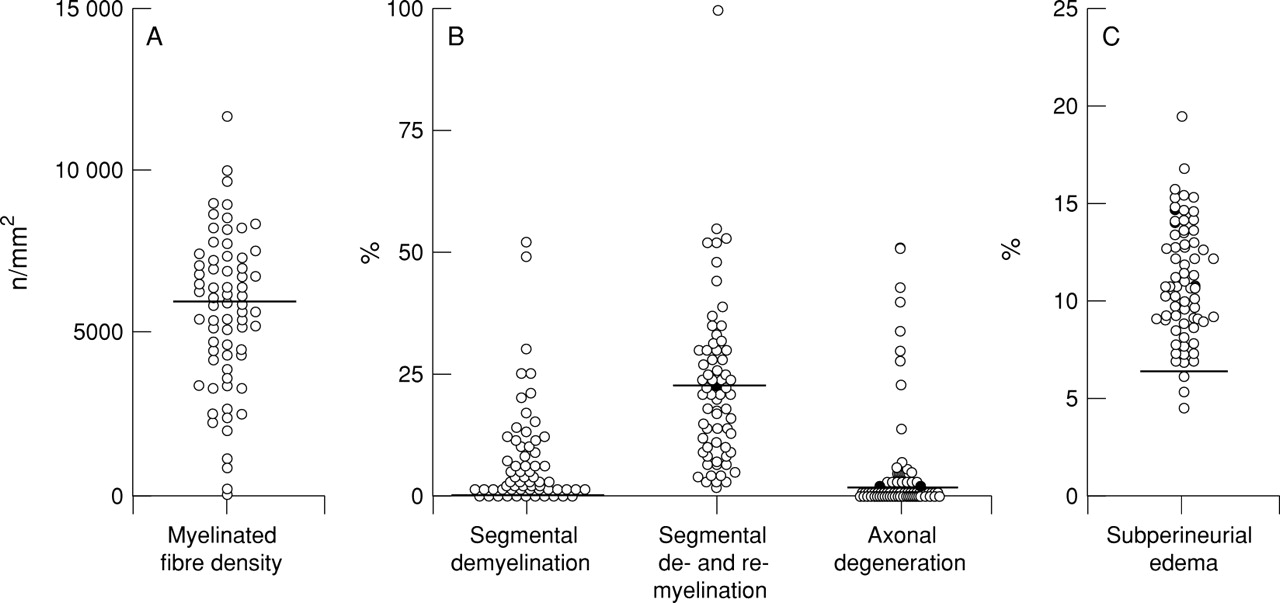
The myelinated fibre density of the sural nerves was diminished in varying degrees (mean (SD) 5679 (2370) /mm2; significantly less than control values, 8679 (1336) /mm2; p<0.0001; fig1A. The incidence of abnormalities in the teased fibre preparation was also increased to various degrees (fig 1 B): 0 to 52% for segmental demyelination (mean (SD) 7.2 (10.6)%); significantly greater than controls 0 (0)%; p<0.0005; 0 to 100% for segmental demyelination and remyelination (mean (SD) 23.0 (16.6)%); significantly increased compared with controls, 9.5 (8.8)%; p<0.05. The extent of active axonal degeneration in CIDP varied widely, ranging from 0 to 51%, (mean (SD) 5.5 (11.1)%), and 23% of the nerves showed values exceeding the mean+2 SD control level (controls, mean (SD) 1.7 (1.4)%). The extent of subperineurial oedema in CIDP (mean (SD), 11.0 (3.1)%) was significantly greater than in controls (mean (SD), 4.6 (1.0)%, p<0.0001; fig 1C). The density of myelinated fibres was significantly inversely correlated with the extent of segmental demyelination and remyelination (fig 2A), and with the extent of segmental demyelination and remyelination plus axonal degeneration (fig2C). Patients with a marked increase in axonal degeneration were also accompanied by a considerable myelinated fibre loss (fig 2B). Myelinated fibre loss was directly correlated with the duration of illness (fig 3), and severe fibre loss (<2000/mm2 in remaining fibres) was found in some patients with a duration of illness exceeding 5 years before the nerve biopsy. Myelinated fibre loss was not significantly correlated with the extent of mononuclear cellular infiltrates as assessed by immunohistochemistry. Infiltrates of LCA positive cells were found using immunohistochemistry in the endoneurium or epineurium in 72% of patients with CIDP. Infiltrates of CD4 positive cells were found in 37%, CD8 positive cells in 35%, and CD68 positive cells in 67% of the nerves from patients with CIDP.
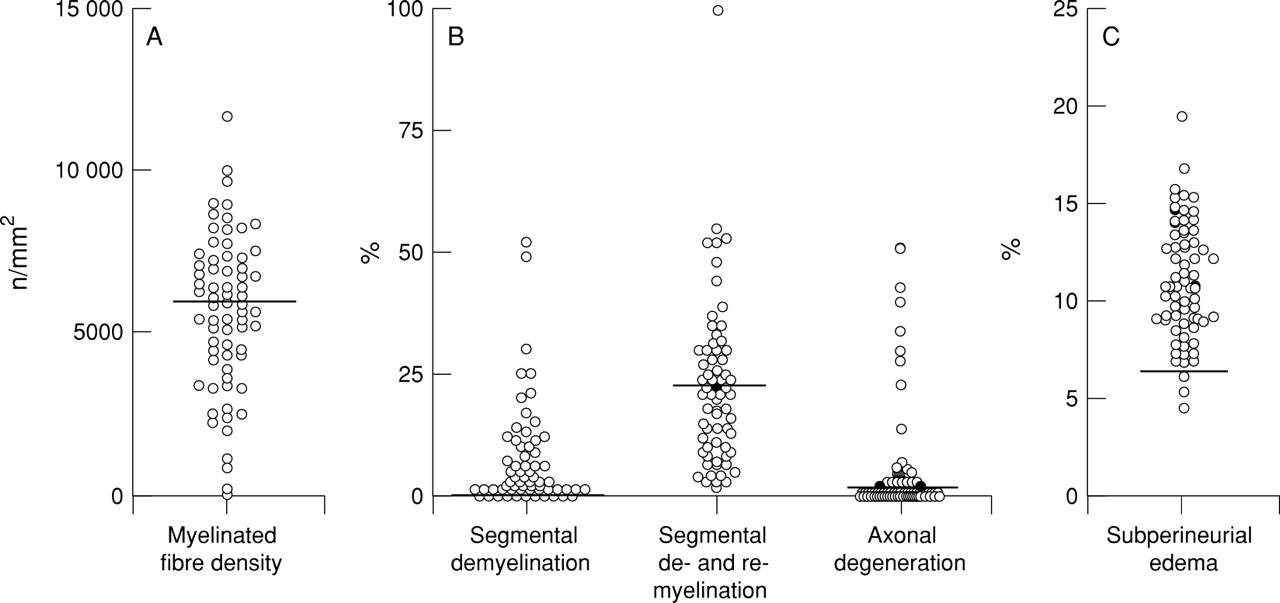
Myelinated fibre pathology in the sural nerves from 71 patients with CIDP. (A) Density of myelinated fibres. The bold line is the mean−SD value in control nerves. The mean value in CIDP is significantly decreased compared with controls (p<0.0001). (B) Teased fibre findings. The bold line is the mean+SD value in control nerves. Frequency of segmental demyelination and segmental demyelination and remyelination was significantly increased compared with control values (p<0.0005 and p<0.05, respectively). (C) Subperineurial oedema. The bold line is the mean+SD value in control nerves. The mean value in CIDP is significantly increased compared with controls (p<0.0001).
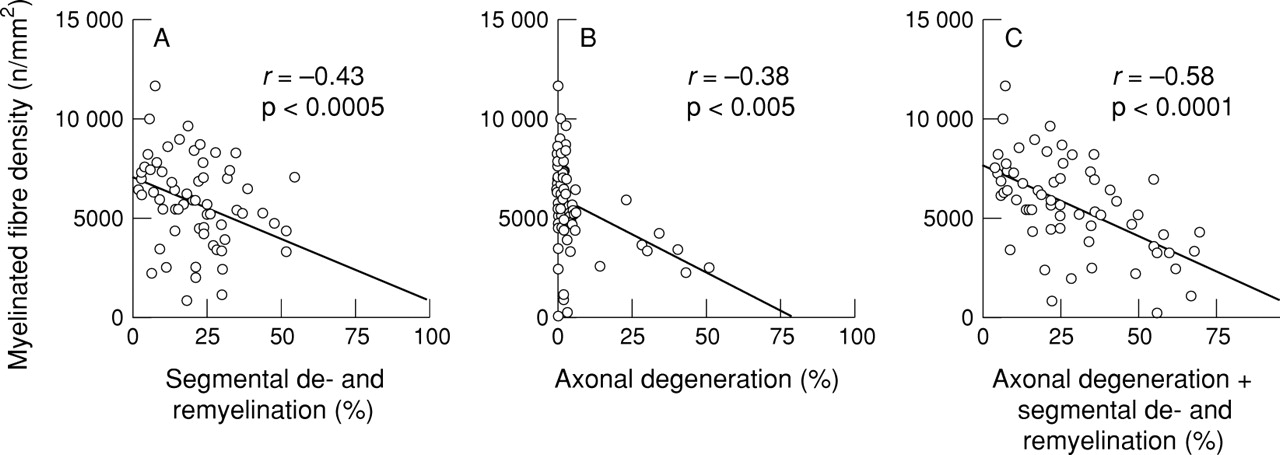
Correlation between myelinated fibre density and abnormalities on teased fibre studies in 71 patients with CIDP. (A) Segmental demyelination and remyelination. (B) Axonal degeneration. (C) Segmental demyelination and remyelination plus axonal degeneration. Myelinated fibre density is inversely correlated with demyelinating changes.

Correlation between myelinated fibre density in the sural nerves and duration of illness in 71 patients with CIDP. A significant inverse correlation is seen.
SPINAL CORDS
The mean number of large motor neurons in the unilateral L4 ventral horn in CIDP (mean (SD) 466 (99)/50 sections) was significantly diminished compared with that of age matched controls (mean (SD) 632 (55)/50 sections, p <0.005; fig 4 and 5 A). The extent of loss of large motor neurons in CIDP was highly variable, the average value being 73.7% of the controls, and the minimum value only 46% of the mean control value (fig 4 and 5A). The small neurons were also significantly depopulated in CIDP, although to a lesser extent (fig5C). The number of medium sized neurons was not significantly decreased (fig 5B). The frequency of ventral horn neurons exhibiting central chromatolysis and accumulations of pNFH was significantly higher in CIDP than in controls (p<0.01 and p<0.05, respectively; fig 6 and table 2). Reactive astrogliosis in the ventral horn was also more prominent in CIDP (table 2). Neuronophagia was occasionally demonstrated by immunohistochemistry using CD68, but lymphocytes positive for CD4, CD8, or UCHL1 were not found in the ventral horns of CIDP. Varying degrees of myelinated fibre loss in the dorsal columns were seen at the L4 level in five of nine patients (table2).

The ventral horn of the L4 segment of the spinal cord from a control (A) and from a patient with CIDP (B). Loss of ventral horn cells, especially large neurons, is evident in CIDP. Klüver-Barrera stain. Bar=100 μm.

The population of large (A), medium sized (B), and small (C) spinal motor neurons in 50 sections of the left L4 segment of the spinal cord. Numbers of large neurons are significantly reduced in CIDP compared with age matched controls (p<0.005). Small neurons are also decreased slightly but significantly (p<0.01).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The ventral horn of the L4 segment spinal cord from a control (A and C) and a patient with CIDP (B and D). A and B, Klüver-Barrera stain; C and D, immunohistochemistry using monoclonal antibody Ta-51, recognising phosphorylated epitope of the high molecular weight subunits of neurofilaments (pNFH). Central chromatolysis (arrowheads in B) and accumulation of pNFH (arrowheads in D) in the cell bodies of spinal motor neurons are evident in the patient with CIDP. Bar=50 μm.
Pathological features of the spinal cord (L4) in patients with CIDP
Discussion
Our study showed that myelinated nerve fibre loss is common in CIDP, and that it is correlated with the extent of demyelination and remyelination on teased fibre analysis as well as with the duration of illness. These findings indicate that both the intensity of the inflammatory demyelinating process and the longevity of the disease process are the factors influencing the severity of myelinated fibre loss.
The precise mechanism of nerve fibre loss in CIDP is unknown. In Guillain-Barré syndrome, axonal degeneration has been found in addition to macrophage mediated segmental demyelination in severe nerve lesions.19 Immunohistochemical study showed antibody and complement mediated attacks on the axolemma of nerve fibres in a Chinese series of axonal Guillain-Barré syndrome,20suggesting that axonal damage can occur as a primary process. However, because antibody and complement deposits in nerves from patients with CIDP are rare,21-26 axonal injury and subsequent fibre loss may be caused by a different mechanism from that suggested in the Chinese series of axonal Guillain-Barré syndrome.
In experimental allergic neuritis (EAN)27-30 and chronic EAN,31-33 models of acute and chronic inflammatory demyelinating neuropathy, axonal degeneration has also been found. Hahnet al and Madrid and Wisniewski reported that the extent of axonal degeneration in nerve roots corresponded to the degree of inflammation and demyelination in their models of EAN.28-30 Moreover, Said et alalso documented axonal degeneration distal to the demyelinated nerve segments by the intraneural injection of antiserum from EAN rabbits.34 Soluble factors such as proteases, phospholipases, lymphotoxins, or tumour necrosis factor- α (TNF-α) from infiltrating mononuclear cells are supposed to be responsible for axonal degeneration in EAN.28 29 35 36 Meanwhile, in biopsied nerves from patients with CIDP, macrophages express TNF-α when attached to myelinated fibres.37 Some of the infiltrating macrophages in the endoneurium as seen in the present study might contribute to axonal damage by releasing soluble factors such as TNF-α.
In our study, the duration of illness is another factor influencing the extent of myelinated fibre loss. Severe myelinated fibre loss in the sural nerves was found in patients with CIDP associated with marked onion bulb formation,38 suggesting that the longstanding, repeated inflammatory demyelinating process is related to myelinated fibre loss. On the other hand, myelin regulates axonal properties such as the focal number, spacing, and phosphorylation level of neurofilaments, axonal calibre, and slow axonal transport.39 40 Therefore, longstanding, repetitive, or persistent demyelination itself might play some part in the process of axonal damage by influencing axonal properties. Actually, hereditary demyelinating neuropathies with abnormal myelin protein genes such as Charcot-Marie-Tooth disease type 1A and 1B, and Déjèrine-Sottas disease are often accompanied by a considerable degree of myelinated fibre loss during a lengthy process.41
The most striking finding in our study was the spinal motor neuron loss in CIDP, which was extensive in some patients. The average loss of one fourth of the large spinal motor neurons, corresponding to α-motor neurons, with a maximum loss of one half in our necropsied series, is more severe than so far inferred. The loss of spinal motor neurons was also evidenced by astrogliosis occurring in the lateral and medial nuclei of the ventral horn, particularly when the loss of these neurons was extensive. Muscle weakness and atrophy were pronounced in patients with extensive spinal motor neuron loss (table 1). This finding indicates that the residual disability in patients with CIDP, especially muscle wasting, is in some way related to spinal motor neuron loss.
Loss of large motor neurons has been reported a long time after limb amputation in humans and cats,42-44 suggesting that longstanding axonal damage may secondarily induce spinal motor neuron loss. Nerve cells are also supposed to be lost when axonal damage occurs close to the cell body.19 Loss of large motor neurons in the spine has been found in necropsied patients with Guillain-Barré syndrome accompanying marked proximal axonal involvements.15 In our series of CIDP, the absence of inflammatory lymphocytic infiltrates in the ventral horn suggests that the ventral horn is not the primary site of inflammation, and that longstanding or proximal motor axonal damage might cause spinal motor neuron loss. An increased rate of central chromatolysis in the spinal motor neurons also suggests the axonal involvement of motor nerves. In addition, an increased accumulation of pNFH in the perikarya of spinal motor neurons may also support this view. pNFH accumulation in the spinal motor neurons has been shown in a wide variety of pathological conditions including amyotrophic lateral sclerosis,14 17toxic neuropathies,45and experimental nerve crush.46 In most cases, a certain degree of impairment of axonal transport is speculated to underlie pNFH accumulation.
Small neurons in the intermediate zone of the ventral horn, corresponding to interneurons, were mildly but significantly decreased in CIDP. The background mechanism of this loss of small neurons is uncertain. Suzuki et al 43 have reported a decrease in small neurons in the cervical intermediate zone as well as in large neurons on the opposite site in a necropsy case studied years after a proximal amputation of one arm, suggesting that contralateral interneurons may be damaged by transneuronal degeneration after loss of large neurons. Small neurons in the intermediate zone in our patients with CIDP might have been lost by a similar mechanism. However, as the degree of loss of small neurons in patients with CIDP was mild, this finding should be confirmed by studying additional cases of CIDP.
In summary, we showed that loss of spinal motor neurons is common in CIDP, and that substantial neuron loss may occur in some cases. Myelinated nerve fibre loss also occurred and was correlated with the extent of segmental demyelation and remyelination, and the duration of illness. These findings are important regarding the long term functional prognosis of CIDP. Ultimately, prevention of these neuronal and axonal losses would be another therapeutic goal in CIDP.
Acknowledgments
This work was performed in collaboration with Drs T Ando and T Yanagi (Department of Neurology, Nagoya Daini Red Cross Hospital, Nagoya), Drs E Isozaki and R Okiyama (Department of Neurology, Tokyo Metropolitan Neurological Hospital, Tokyo), Drs N Takahashi and K Kono (Department of Neurology, Toki General Hospital, Toki), Drs Y Washimi and C Mabuchi (Department of Neurology, Nagoya Ekisaikai Hospital, Nagoya), Drs K Mano and H Watanabe (Department of Neurology, Nagoya Daiichi Red Cross Hospital, Nagoya), Dr K Mokuno (Department of Neurology, Toyohashi Municipal Hospital, Toyohashi), Drs M Konagaya and Y Matsuoka (Department of Neurology, Suzuka National Hospital, Suzuka), Drs T Yasuda and N Murakami (Department of Neurology, National Sanatorium Higashi Nagoya Hospital, Nagoya), Dr E Mukai (Department of Neurology, Nagoya National Hospital, Nagoya), Dr T Sakakibara (Department of Neurology, Chubu Rosai Hospital, Nagoya), Dr T Kameyama (Department of Neurology, Gifu Prefectural Tajimi Hospital, Tajimi), Dr S Ikeda (Department of Medicine, Shinshu University School of Medicine, Matsumoto), Dr A Kawaoi (Department of Pathology, Yamanashi Medical School, Nakakoma), Dr M Sasahara (Department of Pathology, Shiga University of Medical Science, Otsu). We are grateful to Dr J Q Trojanowski (Departments of Pathology & Laboratory Medicine, University of Pennsylvania School of Medicine) for providing the monoclonal antibody Ta-51. We are also grateful for the expert technical assistance of Ms Sugiko Yokoi. This work was partly supported by grants from the Ministry of Welfare and Health of Japan, and by a Centre of Excellence grant from the Ministry of Education, Science, and Culture of Japan.