Article Text

Abstract
OBJECTIVE To estimate the prevalence of visual field defects in patients taking the anticonvulsant drug vigabatrin and to characterise the features of visual dysfunction found.
METHODS Thirty three unselected patients attending neurology and epilepsy clinics were identified as taking vigabatrin and asked to attend for neuro-ophthalmic evaluation. A control group of 16 patients with epilepsy unexposed to vigabatrin was also evaluated. Visual fields were examined by static perimetry using a Humphrey field analyser. Patients underwent detailed ophthalmic examination, various blood tests, and brain MRI where necessary. Visual evoked responses (VERs), electro-oculograms (EOGs), and electroretinograms (ERGs) were recorded.
RESULTS Of 31 assessable patients treated with vigabatrin, 16 (52%) had definitely abnormal visual fields, nine (29%) had fields that were inconclusive, four (13%) had normal fields, and two (6%) proved unable to cooperate with testing. In four patients some plausible cause was found for the field abnormality leaving 12 patients (39%) in whom a definite bilateral field defect was found, possibly caused by vigabatrin treatment. Of 16 control patients none had definitely abnormal fields, 12 (75%) had normal fields, and four (25%) had fields that were inconclusive. The field defects associated with vigabatrin treatment showed a characteristic pattern of concentric peripheral field loss with temporal and macular sparing. The VERs and ERGs were normal. The EOG Arden Index was reduced in patients taking vigabatrin, although this returned towards normal when vigabatrin was stopped, even in the presence of persistent field defects. Multifocal ERGs recorded in two patients were abnormal, showing marked reduction in amplitude of the peripheral focal ERG.
CONCLUSIONS Treatment with vigabatrin was associated with a high prevalence of peripheral visual field defects. This seemed to be the result of a toxic effect of vigabatrin on the retina and seemed to persist if the drug was withdrawn.
- vigabatrin
- visual field
- retina
Statistics from Altmetric.com
Vigabatrin was the first of the novel anticonvulsant drugs to be introduced into clinical practice, in the mid-1980s. It has proved to be a successful and well tolerated drug used for the treatment of epilepsy of partial onset and for infantile spasms (West's syndrome). Chemically vigabatrin resembles γ-aminobutyric acid (GABA), a major inhibitory transmitter in the brain and retina, and is thought to exert its anticonvulsant effect by irreversible inhibition of the enzyme GABA transaminase, which catalyses the inactivation of GABA. Occasional reports had linked vigabatrin treatment with the appearance of visual field defects1 2 and in 1997 we presented three cases of severe persistent visual field constriction in patients who had been treated with vigabatrin.3 Since then several further cases have been reported.4-19
Electrophysiological evidence suggests that the cause of the visual field defects might be a toxic effect of vigabatrin on the retina.3-5 8 9 12-14 17 18 However, reports of visual dysfunction in patients with epilepsy treated with other anticonvulsant drugs20-24 raise the possibility that such deficits might be a relatively common side effect of anticonvulsant treatment or even a feature of the natural history of epilepsy itself. The purpose of this study was to investigate the prevalence of visual field defects in patients treated with vigabatrin and to characterise the features of the visual dysfunction found.
Patients and methods
Patients were recruited from the neurology and epilepsy clinics at Leicester Royal Infirmary and Kettering General Hospital. A total of 33 patients taking vigabatrin were identified and were requested to attend for examination. In two patients, the diagnosis of epilepsy was thought to be incorrect (patients 9 and 10). One patient (patient 1) included in this series has been described (as case 2) elsewhere.3Sixteen patients with epilepsy who had never been exposed to vigabatrin acted as a control group.
Visual fields were initially assessed by threshold related gradient adapted suprathreshold static perimetry using the Humphrey field analyser (Humphrey Systems Inc, San Leandro, CA, USA). In most cases, the 120 point screening program using the threshold related strategy was performed initially. This test identifies locations at which the subject's detection threshold is at least 6 dB more than predicted. The 120 stimulus locations are situated within an eccentricity of 50° nasally, 60° temporally, 40° superiorly, and 55° inferiorly. Patients in whom the screening fields were considered to be abnormal were assessed further in the neuro-ophthalmic clinic at Leicester Royal Infirmary. Each patient underwent detailed ophthalmic examination including direct and indirect ophthalmoscopy by an ophthalmologist after pupillary dilatation, slit lamp examination, and measurement of intraocular pressure. Colour vision was assessed using Ishihara plates. The following blood tests were performed: full blood count, plasma viscosity, urea and electrolytes, glucose, liver function tests, angiotensin converting enzyme, immunoglobulins and serum electrophoresis, C reactive protein, antinuclear factor, vitamin B12, thyroid function tests, and lipids. Brain MRI was performed unless recent brain MRI or CT were avaiable.
The follow up visual field examination comprised threshold static perimetry with the Humphrey field analyser using either program 24–2 or 30–2 which examine 56 and 76 locations out to 21° and 27° respectively and either the full threshold or FASTPAC strategies. Wherever possible, the peripheral field (from 30° to 60° eccentricity) was assessed using program 30/60–2. For central field examination, all patients were fully corrected in trial lens form for the viewing distance of the perimeter. The particular testing regime was tailored to individual patients, bearing in mind the pattern of abnormality identified in the initial screening test and ability to cooperate with the requirements of the tests. All patients with abnormal threshold fields were tested on at least three separate occasions and most patients' fields have been assessed between five and eight times.
All visual field records were assessed blind by one of us (JW) with much experience of automated perimetry. Automated static threshold perimetry is a demanding visual task particularly in patients in whom any cognitive impairment exists. A high standard of reproducibility and reliability was applied before the patients' fields were classified as definitely abnormal. Patients who exhibited visual fields that manifested false responses to the catch trials that were greater than the standard accepted criteria for normality (>20% fixation losses, >33% false negative responses, >33% false positive responses) were deemed unreliable. Patients whose fields manifested so high a degree of threshold variability within test and between test as to render accurate interpretation of the field impossible were deemed inconclusive. It is possible that some patients with “true” visual field defects may have been included in these two groups. However, this policy was adopted so that we could be confident that our estimate of the prevalence of visual field defects in patients taking vigabatrin should be an underestimate rather than an exaggeration.
Routine electrophysiological examinations were performed in the Medical Physics Department at Leicester Royal Infirmary and comprised electrooculograms (EOGs), flash electroretinogram (ERGs), and visual evoked response (VERs), using standard methods. The ERGs were recorded using either a silver/silver chloride skin or a corneal gold foil electrode referred to a silver/silver chloride electrode at the outer canthus of the ipsilateral eye. Five minutes of dark adaptation occurred before the scotopic stimulus was presented (maximal rod/cone response). The amplitudes of the a waves and b waves were measured. Oscillatory potentials were not assessed. The EOG Arden Index was calculated from the minimal potential during the dark adaptation period (dark trough) and the peak amplitude during light adaptation (light peak). The VERs were recorded in response to flash stimulation and to pattern stimulation using a checkerboard alternating at 2 Hz.
Results
PREVALENCE OF VISUAL FIELD DEFECTS IN PATIENTS TREATED WITH VIGABATRIN
Of the 33 patients, two failed to attend, leaving 31 assessable patients. Of these 16 (52%) had visual fields in each eye that were considered definitely abnormal, nine (29%) had fields that were either unreliable or inconclusive, four (13%) had normal fields, and two (6%) proved unable to cooperate with testing. In four of the 16 abnormal cases, some plausible cause was identified for the visual field defect (optic disc drüsen in one, cerebral lesions in two, prior chiasmal compression in one). These four patients were not analysed further. This left 12 patients (39%) in whom a definite bilateral field defect was identified that could not be explained in terms of any other ophthalmic or neurological pathology. Demographic and clinical details of the 25 patients analysed fully are provided in table 1, drug histories in table 2, and neuro-ophthalmic findings in table 3.
Demographic and clinical details of patients treated with vigabatrin
Drug history of patients treated with vigabatrin
Neuro-ophthalmic findings in patients treated with vigabatrin
In the control group of 16 patients, 12 (75%) had normal fields whereas four (25%) had fields that were considered inconclusive. None of the control patients had fields that were considered definitely abnormal. Demographic and clinical details of the control group are provided in table 4.
Demographic and clinical details of control patients
The 12 patients with field loss linked to vigabatrin (defect group) comprised five men and seven women with a mean age of 32.8 years. The patients treated with vigabatrin and with normal fields (no defect group) comprised two men and two women with a mean age of 36.3 years. Two of the four patients in the no defect group were of Asian descent; all patients in the defect group were of European origin. The duration of epilepsy in the defect group had a mean of 22.3 years whereas that of the no defect group was 25.5 years. The patients in the control group had a mean age of 37.3 years and their duration of epilepsy had a mean of 14.3 years. The mean maximum daily vigabatrin dose of the defect group was 3.1 g (range 1.5–5.0 g) with a mean duration of treatment before initial assessment of 4.7 years (range 1.7–8.2 years). The mean maximum dose of the no defect group was 3.0 g (range 2.0–4.0 g) and the mean treatment duration was 2.6 years (range 0.8–5.3 years). An attempt was made to calculate cumulative vigabatrin doses for all patients. In the defect group the mean cumulative vigabatrin dose was 4.4 kg (range 1.3–8.9 kg). In the no defect group the mean cumulative dose was 1.7 kg (range 0.7–2.5 kg). Both partial onset and idiopathic generalised epilepsies were represented in all groups. Two patients in the defect group (patients 9 and 10) did not have epilepsy. All groups had been exposed to a wide variety of other anticonvulsant drugs.
Four patients in the defect group had optic discs in each eye that were considered to show definite pallor, whereas five patients had slightly pale discs and three patients were normal. In the no defect group slight pallor was recorded in two patients and two were normal. In the defect group the peripheral retina seemed hypopigmented with an easily visible choroidal circulation in two patients (tigroid fundus), but was normal in the remaining 10 patients and in all four of the no defect group. Visual acuity was in the normal range for all but two patients (patients 6 and 19; best acuities 6/12) and no abnormality of colour vision was detected using Ishihara plates in all but one patient (patient 1) who was a congenital protanope.
Despite the presence of definite field defects, three of the 12 patients in the defect group had no visual symptoms of any kind. Five patients reported mild symptoms of blurring or phosphenes and four reported more severe symptoms (marked blurring in one patient and tunnel vision in three patients). The patients with severe symptoms were those with the most advanced field defects.
CHARACTERISTICS OF VISUAL FIELD DEFECTS ASSOCIATED WITH VIGABATRIN TREATMENT
In all patients with definite field defects, the abnormalities were symmetric between the two eyes of a given patient, were most marked beyond about 15–21° eccentricity, and were often absolute. The most advanced field defects were concentric, but in milder cases the field loss was proportionately more extensive in the nasal field both in terms of area and depth resulting in a characteristic pattern of binasal field loss extending in an annulus across the horizontal midline with a tendency for sparing of the temporal field (fig 1).
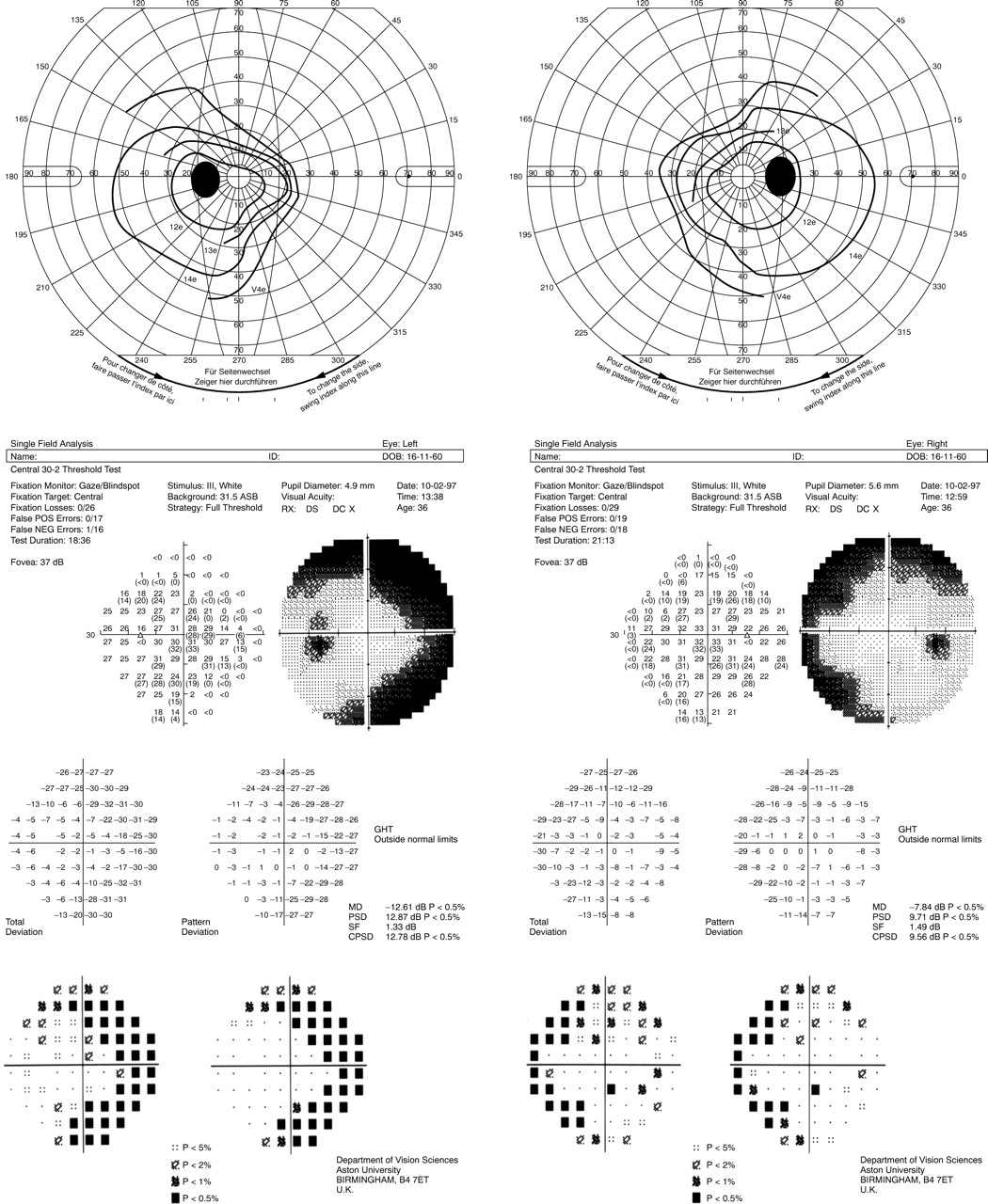
Goldmann kinetic visual field (top) and Humphrey field analyser program 30–2 full threshold static visual field (bottom) for patient 5.
ELECTROPHYSIOLOGICAL CONSEQUENCES OF VIGABATRIN TREATMENT
Using conventional electrophysiological measures of visual function (ERG, EOG, and VER), the only abnormality that seemed strongly associated with vigabatrin use was reduction of the Arden Index of the electro-oculogram (EOG). All other results (the scotopic and photopic ERG amplitudes and the VER latencies) were usually within normal limits. The Arden Index was significantly reduced in patients who were taking vigabatrin at the time that the EOG was assessed (fig 2A). Patients who had previously been exposed to vigabatrin, but had discontinued the drug before EOG testing, showed results that often lay within the normal range. Control subjects taking other anticonvulsant drugs, but never exposed to vigabatrin, showed EOG Arden Indices that were significantly higher than those found in the patients who had discontinued vigabatrin. In eight patients, EOGs were recorded before and several months after discontinuing vigabatrin. In all patients, the EOG Arden Index increased substantially on discontinuation (fig2B).

(A) Results of the EOG Arden Index for (1) patients recorded while taking vigabatrin, (2) patients recorded after vigabatrin discontinuation, (3) patients on other anticonvulsant drugs unexposed to vigabatrin. The horizontal lines represent −1 and −2 SD below the mean for EOGs recorded in our laboratory. (B) EOG Arden Index results for those patients in whom recordings were made both before and after vigabatrin was discontinued.
In view of the findings from conventional electrophysiology and to confirm that the visual field defects were in fact related to retinal abnormality multifocal ERGs were performed on two patients (patients 1 and 5) using the VERIS ™ system (Tomey). This system uses discrete stimulation of 103 hexagonal elements within a visual field subtending 50° horizontally and 40° vertically. The 103 elements are scaled with eccentricity to elicit responses of about equal amplitude. Each element is pseudorandomly stimulated achromatically and the responses to each specific element separately stored and averaged.25
The two patients showed a marked overall reduction in amplitude of the focal ERG peripherally with evidence of macular sparing. Data for patient 5 are shown in fig 3 and comparison with that patient's visual fields (fig 1) shows a similar pattern of predominantly binasal abnormality. Despite previous findings of increased latency of the b wave of the photopic ERG in patients receiving vigabatrin26 there was no evidence of peripheral specific delay in b wave latency in the multifocal ERG.

{kind=link}
{kind=link}
{kind=link}
Representation of VERIS responses (nV) in patient 5 plotted according to the field hexagon that elicited the response. White areas indicate values within normal limits (±2 SD), grey areas indicate abnormally low responses beyond 2 SD from the normal and black areas indicate abnormally low responses beyond 3 SD from the normal.
Discussion
These results suggest that vigabatrin treatment is associated with peripheral visual field defects in a substantial proportion of patients. It has been suggested that field defects may arise as a class effect of anticonvulsant drugs or as a feature of epilepsy itself.4 We are aware of only one study that attempted to assess the prevalence of visual field defects in patients with epilepsy.20 The prevalence of field defects was high (20% of 55 patients), but this patient group was selected by occipital EEG foci and many had structural lesions in the occipital lobes. Figures derived from this atypical group cannot be applied to the whole epileptic population. Rare cases have been reported of visual field defects associated with other anticonvulsant drugs. One report described constricted visual fields caused by phenytoin, but this was the result of prolonged toxic blood concentrations in a patient with a rare defect of drug metabolism.21 Another report implicated oral diazepam22 taken in large doses (100 mg/day) as an anxiolytic agent, but the importance of this to epilepsy practice is not clear. A recent report described persistent visual field constriction after 11 years of treatment with the GABA agonist drug progabide.24 This is of particular interest as it raises the possibility that visual field constriction may be a class effect of drugs affecting GABAergic mechanisms. In our control group no definite field defects were found. No attempt was made in this study to match vigabatrin treated and control patients for age, sex, type, and duration of epilepsy. A study comparing patients treated with vigabatrin and carefully matched controls is under way and will be reported in due course.
If the visual field defects reported here were caused by vigabatrin, what is likely to be the mechanism underlying their production? It seems probable that the site of damage is the retina. Concentric peripheral visual field loss is most commonly reported in retinal disease and is unlike the patterns usually found in optic nerve disorders. Most optic neuropathies result in loss of visual acuity, defective colour vision, and abnormal VERs, which were spared in our patients. In addition, two electrophysiological measurements of retinal function, the EOG and multifocal ERG, were abnormal in many of our patients. We find these arguments that vigabatrin causes retinal malfunction persuasive.
The EOG abnormality was a reduction of the Arden Index. This is the ratio of EOG amplitude in light and dark adapted conditions and is a measure of retinal pigment epithelial function. This abnormality is not directly related to the cause of the field defect as it tends to recover when vigabatrin is withdrawn, whereas the field defects persist. The multifocal ERG abnormalities described above suggest photoreceptor malfunction, although their correlation with field loss was poor. It seems likely that multifocal ERG abnormalities lie closer to the cause of field loss than those of the EOG as they persist after drug withdrawal. A recent study13 suggested that the amplitude of the photopic ERG was reduced by vigabatrin, with similar reduction in amplitude of oscillatory potentials. These patients were receiving vigabatrin at the time of investigation and these abnormalities have been shown26 27 to be associated with current vigabatrin therapy. Comprehensive electrophysiological assessment of a further patient showed a normal flash ERG, although with diminished oscillatory potentials, and an alteration in the relative amplitudes of the P50 and N95 components of the pattern ERG when the stimulus was displayed within the affected field.18 Multifocal ERG showed no loss of amplitude (by contrast with our findings) although the shape of the waveform was altered in the areas of affected field. The VER and EOG results were normal in this patient, although it may be that vigabatrin had been withdrawn by the time that the EOG was recorded. The authors suggested that an alteration of inner retinal function was the cause of the field defect.
Toxicological studies on vigabatrin have concentrated on microvacuolation in myelin sheaths in the white matter of rats, mice, and dogs.28-30 No such lesions have been found in monkeys, nor in those human cases subjected to necropsy.31Studies of evoked potential latencies in patients taking vigabatrin have provided no evidence of demyelination,32 nor was any prolongation of VER latencies seen in our patients. A recent case report described bilateral optic neuropathy in a child taking vigabatrin in whom electrophysiological investigations suggested optic nerve demyelination.33 The clinical features of this patient do not resemble those in our series and it is possible that this was a case of idiopathic optic neuritis associated with vigabatrin therapy by chance. We do not think that damage to myelin underlies the field defects that we have recorded. The only toxicological research into vigabatrin's effect on the retina of which we are aware showed that in albino but not in pigmented rats, vigabatrin had a dose dependent effect on the outer retina characterised by disruption of the outer nuclear layer,34 which contains the photoreceptor nuclei.
GABA is known to have a neurotransmitter role in horizontal and amacrine cells of the vertebrate retina. GABAergic transmission connects horizontal cells to bipolar cells, to other horizontal cells, and to photoreceptors.35 Perfusion with GABA agonists alters the responses of horizontal, amacrine, and ganglion cells in the cat retina.36 Vigabatrin causes GABA to accumulate in retinal glial cells in rats.37 Subcutaneous vigabatrin produced greater inactivation of GABA transaminase in the retina than in the brain, leading to the suggestion that the drug may enter the retina selectively via the aqueous humour.38 In other studies tolerance to higher vigabatrin doses was found in rat brain and spinal cord, but not in the retina.39 For these reasons, vigabatrin may be particularly likely to produce retinal side effects.
In most cases our patients' field defects were asymptomatic or produced mild symptoms. Only four of the patients (13%) had appreciable visual symptoms. Whether asymptomatic field defects are of clinical importance depends on the likelihood of further progression if treatment is continued. The fact that many of our patients were unaware of impairment of the peripheral visual field does not detract from their potential importance. It is known that patients with chronic glaucoma will tolerate extensive loss of peripheral visual field without complaint if that loss is gradual. Field loss in the binasal distribution typical of vigabatrin is especially likely to go unnoticed as the loss of nasal sensitivity in one eye will be compensated by the relatively preserved temporal field in the other eye.
The field defects found in our patients persisted when vigabatrin was withdrawn, although modest improvement was seen in three. However, the existence of a perimetric learning effect, whereby sensitivity improves with repeated examination, is well documented. Patients with severe defects have shown no improvement and remain symptomatic 3 years after vigabatrin withdrawal. Two of our patients with field defects are still taking vigabatrin as the beneficial effect of the drug was thought to outweigh the risk of progression of currently asymptomatic visual field loss. In patient 4 no progression of the field defect has been seen over a period of 39 months. In patient 8 there seems to have been minor deterioration of his visual fields over a period of 11 months. Those patients with possible field defects whose data were inconclusive have shown no tendency to deteriorate over periods of up to 2 years. Our patients with symptomatic field defects agree that their visual deterioration was insidious, but the question whether asymptomatic field defects will gradually deteriorate and become symptomatic remains unanswered. Our data suggest that the cumulative dose of vigabatrin may be a factor in the development of field defects, and therefore we suggest that patients taking this drug should undergo regular visual field assessment for as long as treatment lasts.
References
Linked Articles
- Editorial commentary