Article Text

Abstract
OBJECTIVES To determine the molecular basis for autosomal dominant intermediate hereditary motor and sensory neuropathy (HMSN) in a four generation family. The gene defects in families with intermediate HMSN are not known, but it has been suggested that most have X linked HMSN.
METHODS All participating family members were examined clinically. Genomic DNA was obtained from 10 affected and seven unaffected members. Linkage analysis for the known HMSN loci was first performed. Mutations in the peripheral myelin protein zero gene (PMP0) were sought in two affected members, using one unaffected member for comparison, by amplification of the six exons of the gene followed by single strand conformation polymorphism (SSCP) analysis, dideoxy fingerprinting (ddF), and sequencing. Subsequently, the mutation was screened for in all affected and unaffected members in the family using Alu I digestion and in 100 unrelated control subjects using “snap back” SSCP analysis. Sequencing of cDNA from a sural nerve biopsy from an affected member was also performed.
RESULTS The clinical phenotype was of variable severity, with motor nerve conduction velocities in the intermediate range. Linkage to PMP0 was demonstrated. Analysis of genomic DNA and cDNA for PMP0 identified a novel codon 35 GAC to TAC mutation. The mutation produces an inferred amino acid change of aspartate to tyrosine at codon six of the processed protein (Asp6Tyr) in the extracellular domain and was present in all affected family members but not in 100 unrelated controls.
CONCLUSIONS The present findings further extend the range of phenotypes associated with PMP0 mutations and indicate that families with “intermediate” HMSN need not necessarily be X-linked as previously suggested.
- intermediate Charcot-Marie-Tooth disease
- myelin protein zero gene
- missense mutation
Statistics from Altmetric.com
Cases of autosomal demyelinating hereditary motor and sensory neuropathy (HMSNI or CMT1) have been associated with duplications of, or point mutations in, the PMP-22 gene on chromosome 17p11.2 (CMT1A),1 2 or, less often, with mutations in the peripheral myelin protein zero gene (PMP0) on chromosome 1q22–23 (CMT1B).3-5 The neuronal form (HMSNII, CMT2) has been linked to loci at 1p35-p36,6 3q13-q22,7 and 7p14,8 but no mutated gene has been identified. Linkage to and mutation of PMP0 have also been reported in families with HMSNII.9-12 Finally, mutations in PMP0 have been described in isolated cases of Dejerine-Sottas disease (HMSNIII)13 14 and congenital hypomyelination.15
The gene defects in families with a clinical phenotype and nerve conduction velocities intermediate between those of HMSNI and II (“intermediate” HMSN) are not known, but it has been suggested that most, if not all, such families have the X-linked form of HMSN (HMSN-X)16 17 which has been associated with mutations in the connexin 32 gene (Cx32) on Xq13.18 19 Linkage to the known HMSNI and II loci was not found in a previously reported Italian family with autosomal dominant intermediate HMSN and it was concluded that the intermediate form of HMSN is a separate entity.20
We report here the finding of a novel mutation in the PMP0 gene in a four generation family of Macedonian origin with autosomal dominant HMSN characterised by a clinical phenotype of variable severity and motor nerve conduction velocities in the intermediate range. Linkage of the disease in this family to the PMP0 region was previously reported in abstract form.21
Methods
CLINICAL STUDIES
The family tree is shown in figure 1. Of ten known affected members, two were deceased. Six of the eight living affected family members were available for clinical examination and nerve conduction studies. In the two who were not available for examination, the diagnosis had been made previously by one of us (BAK). Fourteen asymptomatic family members were also examined. Motor and sensory nerve conduction were measured in the median and ulnar nerves using standard techniques in use in this department.22 A sural nerve biopsy was performed in two affected members (II.6, III.8).

Pedigree of the family, showing the conserved haplotype for loci CRP, APOA2, and D1S104 and the recombinations with D1S305 and D1S194.
MOLECULAR STUDIES
Genomic DNA
Genomic DNA was extracted from peripheral blood from 10 affected and seven unaffected family members.
Linkage analysis
Microsatellite markers were analysed according to standard procedures in this laboratory.23
Exonic amplification
The six exons of PMP0 were amplified individually using the primer pairs listed in table 1. Each 25 μl polymerase chain reaction (PCR) volume consisted of 100 ng of each primer pair, 50 ng genomic DNA, 2 mM MgCl2, 200 μM of each deoxynucleotide triphosphate, 0.55 U Tth Plus DNA polymerase (Biotech International Ltd), and 2.5 μl 10X reaction buffer (Biotech International Ltd). Amplification conditions for all exons were an initial 94°C for 4.5 minutes followed by 35 cycles of 94°C for 30 seconds, 1 minute at 58°C, and 45 seconds at 72°C. Gel purified products of each of the exons were analysed for single stranded conformation polymorphisms (SSCPs).26 27To increase the likelihood of mutation detection, various gel fractionation conditions were used. These included 19:1, 29:1, 37.5:1, and 99:1 polyacrylamide gel ratios, 1:3 and 1:7 DNA to formamide loading buffer (FLB) (deionised formamide, 10 mM NaOH, 0.05% bromophenol blue, 0.05% xylene cyanol) ratios for samples, and electrophoresis at room temperature and 4°C, and at 200 V or 400 V.
Primers used to amplify P zero genomic DNA and cDNA
Dideoxy fingerprinting
Dideoxy fingerprinting (ddF),28 was performed using radiolabelled dideoxy terminators and thermostable sequenase (Amersham Life Sciences). Approximately 100 ng purified PCR product from each exon was reamplified for 55 cycles using the same parameters as above. After mixing the samples with the manufacturer’s stop solution, a 4 μl aliquot was denatured for 3 minutes at 94°C before electrophoresis through a 6%, 29:1 non-denaturing polyacrylamide gel for 2 hours 10 minutes at 35 W. A 1.2 X and 0.8 X TBE buffer concentration was used in the gel and the running buffer respectively. Biomax MR film (Kodak) was exposed overnight with the radioactive gel.
Sequencing
Polymerase chain reaction products for sequencing were purified from unreacted primers and nucleotides using QIAquick PCR spin columns (QIAgen) as described by the manufacturer. The purified products were sequenced using the ABI BigDye™ terminator kit and the same primers as used for the initial exonic amplifications and analysed on an ABI 373A automated sequencer.
Alu I digestion
A forward mismatch primer was designed, 5′-CCCAGGCCATCGTGGTTTACAGC-3′, which would introduce an Alu I enzyme site only if the putative mutation was present. This primer was used with the original exon 2 reverse primer to amplify exon 2 of the genomic DNA using the same PCR conditions as for all the exonic amplifications. The PCR products were ethanol precipitated and then digested with Alu I (Promega) according to the manufacturer’s directions. Digestion products were electrophoresed through a 12%, 29:1 non-denaturing polyacrylamide gel at 250 V, for 1.5 hours at room temperature. Bands were visualised by silver staining.29
Snap back SSCP analysis
A snap back primer30 for exon 2, 5′- ACAC CGACAGTTCTGTTATCCAACCCCAG-3’, was designed to screen 100 unrelated, normal controls. This reverse primer was used with the original exon 2 forward primer to amplify genomic DNA. Apart from the new primer, the previous PCR mixture was used, with amplification conditions at 94°C for 4.5 minutes followed by 38 cycles of 94°C for 30 seconds, 60°C for 45 seconds, and 72°C for 45 seconds. A 3 μl aliquot from each amplification solution was mixed with 9 μl of FLB and denatured at 96°C for 3 minutes before being cooled on ice. Chilled samples were electrophoresed through a 12%, 29:1 non-denaturing polyacrylamide gel at 400 V for 1 hour 45 minutes. The gel system was packed in ice to maintain a temperature of less than 18°C. Bands were visualised by silver staining (fig2).29

SSCP analysis of P zero gene exon 2 in one affected member and four controls.
cDNA analysis
RNA was extracted from the nerve biopsy from patient II.6 using TRI REAGENT R (MRC Inc). Total RNA (2 μg) was used to synthesise first strand cDNA with M-MLV reverse transcriptase (Perkin-Elmer). The P0 coding region was amplified with the cDNA A primer in the 5′ UTR (table 1), and the exon 6 reverse primer in the 3′UTR (table 1). The 25 μl PCR mixture was as for the genomic amplifications above, with about 10% of the total cDNA sample being amplified by a denaturation step at 94°C for 4.5 minutes, followed by 40 cycles of 94°C for 30 seconds, 55°C for 1 minute, and 72°C for 1 minute. The product was sequenced using ABI BigDye™ terminator kits as for the genomic DNA sequencing, with three internal cDNA primers B, C, and D (table 1) as well as those used for the original cDNA amplification.
Results
CLINICAL STUDIES
In addition to the eight known affected members, two of the asymptomatic members (fig 1: IV.6, IV.7) were also found to be affected clinically and had abnormal nerve conduction studies (table2).
Summary of clinical details and motor nerve conduction velocities in eight affected patients
Affected members displayed a symmetric pattern of distal muscle atrophy, weakness, and sensory impairment in the lower limbs and to a lesser extent in the upper limbs, with depression of deep tendon reflexes. Weakness of knee and hip flexors was present in some of the more severely affected members. Sensory involvement was present in five of the eight cases. As shown in table 2, there was considerable variability in the severity of motor and sensory involvement which was most severe in two affected women in their 50s. A mild postural and kinetic upper limb tremor was present in three cases. There was no palpable thickening of peripheral nerve trunks in any of the affected members. As shown in table 2, the motor nerve conduction velocities varied from 24–41 m/s for the median nerve and from 33–48 m/s for the ulnar nerve. Sensory nerve action potentials at the wrist were markedly delayed and attenuated in all affected members.
NERVE BIOPSY
In II:6 there was advanced fibre loss with few residual myelinated fibres. In III:8 there was non-selective drop out of myelinated fibres. Light and electron microscopy and teased fibre preparations showed evidence of axonal degeneration. There was no onion bulb formation. Teased fibre preparations also showed some thinly myelinated internodes suggesting segmental demyelination and remyelination. Neither tomacula nor fibres with uncompacted myelin sheaths were found.
MOLECULAR STUDIES
Linkage studies
The pedigree, with DNA available from 10 affected members (fig 1), was large enough to obtain significant evidence of linkage to candidate loci in the human genome. Linkage analysis was begun with the known loci for CMT1A, the peripheral myelin protein gene (PMP22) on chromosome 17,1 31 CMT1B, the peripheral myelin PMP0,3 5 and CMT2A6 on chromosome 1. Recombinations with markers D17S839, D17S921, and D17S922 within and flanking the CMT1A duplication region on chromosome 17 and two point LOD scores, calculated with the FASTLINK version of MLINK,32-34 of less than −2.00 allowed exclusion of the CMT1A region. The CMT2A locus was similarly excluded. For the CMT1B region on chromosome 1 however, no recombinations were seen with the markers CRP, APOA2, and D1S104 (fig 1). The multipoint LOD score calculated with the FASTLINK version of LINKMAP, using a dominant fully penetrant model, ignoring the unaffected members in generation IV and using a disease gene frequency of 0.0001, was 3.12 at both CRP and APOA2.
Genomic DNA analysis
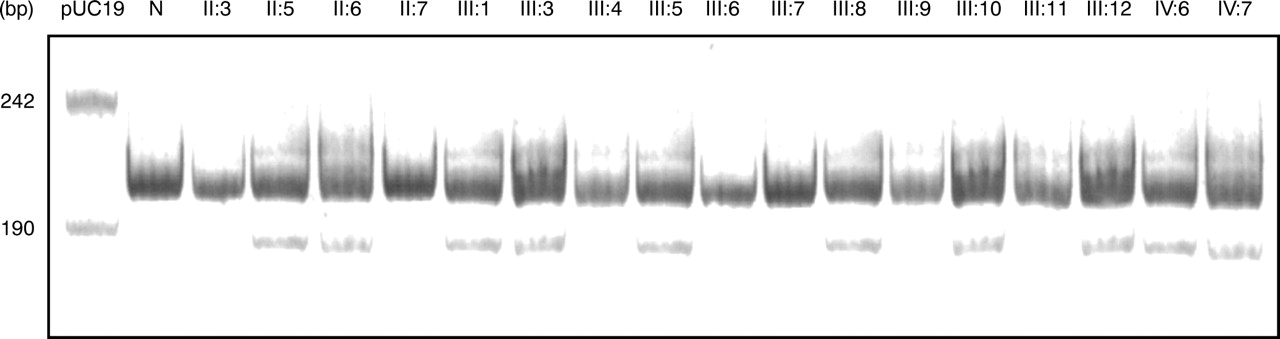
The six exons of PMP0 were amplified from two affected family members and one normal member. The amplicons from affected members were compared with normal controls by SSCP analysis26 27 under a range of conditions, but no polymorphisms were detected. The amplified products from each exon were then analysed by dideoxy fingerprinting (ddF)28 which indicated differently migrating species in exon 2 for the affected samples compared with the normal. Sequencing of the PCR products from exon two of two affected members and one control indicated a heterozygous change from GAC to TAC (aspartate to tyrosine) at codon 35 of the cDNA (numbered according to the Human Gene Mutation Database35) (fig 3). Codon 35 is the equivalent of amino acid 6 of the processed protein15 36 and the inferred amino acid change may therefore be designated Asp6Tyr. Polymerase chain reaction products of exon 2 from all 10 affected members amplified using the mismatch primer which introduces an Alu I site when the mutation is present, showed a smaller, 171 bp product, after Alu I digestion. Polymerase chain reaction products from the unaffected members did not (fig 4). An SSCP was obtained using the snap back primer amplicons in samples from all affected family members, but from none of 100 unrelated, normal controls. Sequencing the PCR products for all other exons from two affected members and one control gave no evidence of any other mutation.

Heterozygote sequencing of exon 2 using automated sequencing. The mutation is seen as a double T and G peak in both cDNA and genomic DNA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Alu I digest of the myelin protein zero gene exon 2 amplicons using the mismatch primer. N = normal control, other lane numbers correspond to the members in the pedigree in fig 1. All affected members show the smaller Alu I digestion product expected from the mutation.
cDNA analysis
Sequencing of the entire coding region of the PMP0 cDNA from one affected member and one control confirmed the presence of the codon 35 mutation at the cDNA level and gave no evidence of any other mutation.
Discussion
The two main types of HMSN (types I and II) have traditionally been differentiated on the basis of reduced motor nerve conduction velocities and pathological evidence of a hypertrophic demyelinating peripheral neuropathy in type I and relative preservation of conduction velocities and axonal degeneration in type II.37-39 A motor conduction velocity in the median nerve of 30 m/s40or 38 m/s41 has variably been suggested as an arbitrary cut off level for separating HMSN types I and II, although it has been recognised that there is a certain amount of overlap between the two types. The nosological status of patients and families with borderline motor nerve conduction velocities in the 30–40 m/s range has been unclear and such families have been referred to as having intermediate HMSN in the literature.42 43 Harding17suggested that most, if not all, such families have the X-linked form of HMSN. In X-linked HMSN, there is considerable variability in clinical and neurophysiological features with, in general, males being more severely affected than females, who may be mildly affected or asymptomatic.44 45 The family reported here was considered to have intermediate HMSN on the basis of borderline motor nerve conduction velocities similar to those in X-linked families. The nerve biopsy results were more consistent with HMSN II. The pattern of inheritance was clearly autosomal dominant and, by contrast with HMSN-X families, two females were more severely affected than the males.
Point mutations in PMP0 were first described in CMT1B by three groups in 19933-5 and subsequently confirmed by other groups.46 Mutations in PMP0 have since been reported in association with a wide range of phenotypes. These have included severe early onset disease conforming to the Dejerine-Sottas syndrome13 14 47 and families classified as HMSN type II with Ser15Phe10 or Thr124Met mutations.11 12 The Asp6Tyr missense mutation found in the present family has not previously been reported but is consistent with previously published PMP0 mutations, most of which have been missense or nonsense substitutions in exons 2 or 3, the portion of the gene coding for the extracellular and transmembrane domains of the protein.17 It is of interest that the mutation Thr5Ile, at a neighbouring amino acid to that mutated in the present study, results in classic HMSN type I, with conduction velocities <10 m/s in the median nerve.48
The finding of a mutation in PMP0 in the present family with intermediate HMSN adds to the range of phenotypes associated with PMP0 mutations. The failure to find a mutation in PMP0 in the intermediate HMSN family reported by Villanova et al 20 indicates that there is genetic heterogeneity in the intermediate form of HMSN as in HMSNI and HMSNII.
Peripheral myelin protein zero is the major structural component of peripheral nervous system myelin, and is an integral membrane glycoprotein belonging to the immunoglobulin superfamily which is encoded by six exons.49 The protein has a homophilic extracellular domain which is thought to act as an adhesion molecule and to play an important role in the compaction of the myelin sheath.49 50 A mutation in the extracellular domain of the protein could therefore interfere with the normal formation and maintenance of the myelin sheath. Meijerink et al 47 found increased numbers of fibres with uncompacted myelin sheaths and evidence of myelin breakdown on electron microscopy in two patients with heterozygous Arg69His and Arg69Cys mutations, whereas Thomas et al 51 found a tomaculous neuropathy in those with identified PMP0 mutations. In the present study, a sural nerve biopsy in two affected persons showed evidence of both axonal degeneration and segmental demyelination with loss of myelinated nerve fibres, but did not show these other changes. There is thus also a considerable variation in the pathological phenotype in those with confirmed PMP0 mutations.
Recent studies employing transfected insect cell lines have indicated a possible correlation between cellular adhesion capability and the severity of the clinical phenotype with different PMP0 point mutations.52 More detailed genotype-phenotype correlation may come from the study of transgenic mice expressing dominant PMP0 mutations.
Acknowledgments
This work was funded by the National Health and Medical Research Council of Australia Project Grant 960592 and the Neuromuscular Foundation of Western Australia. We are grateful to Dr G Danta and Dr A Kermode who performed nerve conduction studies in two of the subjects, and to Dr L Middleton and Professor A D Roses for helpful discussions in the early stages of the study. Mrs S Moncrieff provided secretarial assistance.