Article Text

Abstract
Background: Alzheimer's disease (AD) is characterised by functional impairment, cerebral atrophy, and degeneration of specific neuronal populations, especially pyramidal neurones of the cerebral cortex and hippocampal formation. Although patients with subcortical vascular dementia have been shown to have similar metabolic and volumetric deficits to those with AD, the underlying pathogenesis of these changes is poorly understood.
Objective: To determine whether pyramidal cell loss occurs in small vessel disease (SVD) dementia by quantifying hippocampal volume and CA1 neurone number.
Methods: Fifty four prospectively studied patients with dementia were screened, and four patients fulfilling criteria for SVD with no other significant neuropathological abnormality were identified. These were compared with five patients fulfilling criteria for AD and seven controls matched for age and sex. The hippocampal formation was serially sectioned, and the number of CA1 pyramidal neurones estimated using the optical dissector technique. Analysis of variance was used to evaluate group differences.
Results: Patients in both the AD and SVD groups showed a substantial loss of pyramidal neurones from the CA1 region. The pattern of hippocampal atrophy and the degree of CA1 neuronal loss were similar in the two dementia groups.
Conclusions: These findings support recent in vivo studies showing similar metabolic deficits and atrophy in AD and subcortical vascular dementia. In addition, they provide evidence that the underlying cause of these abnormalities is a similar loss of neurones. Whereas the cause of the neuronal loss in AD is related to the deposition of abnormal proteins, the cause in SVD is unknown. In the absence of other pathologies, damage to cerebral microvasculature should be considered a likely candidate.
- Alzheimer's disease
- SVD, small vessel disease
- dementia
- cerebral atrophy
- neurones
- AD, Alzheimer's disease
- SVD, small vessel disease
Statistics from Altmetric.com
Vascular dementia is recognised as one of the most common causes of dementia after Alzheimer's disease (AD). Traditionally, vascular dementia has been distinguished from AD by the pattern of clinical progression, with vascular dementia characterised by a stepwise rather than a slowly progressive course of deterioration. However, this view appears overly simplistic, as clinical presentations can vary enormously in patients with vascular dementia.1–5 In particular, some cases of vascular dementia are clinically indistinguishable from AD as they show no evidence of a stepwise deterioration.2 At autopsy, these patients have small vessel disease (SVD), leucoencephalopathy, and microscopic infarction rather than the pathology of AD.2 This has been confirmed in a carefully selected autopsy series, leading Esiri and colleagues4 to suggest that microvascular disease, rather than macroscopic infarction, is the most common pathology underlying vascular dementia.
Although SVD is being increasingly recognised as a subtype of vascular dementia,5, 6 no validated criteria for the identification of such cases, either clinically or pathologically, have been established. At present, the pathological classification of SVD is largely descriptive and often relies on the pathologist's opinion of whether the vascular pathology contributed to the dementia.7 The study by Esiri and colleagues4 proposed a semiquantitative grading scale for SVD using the presence and increasing severity of a number of features of microvascular pathology. The systematic use of this scale may lead to a better understanding of the mechanisms by which SVD causes, or contributes to, dementia.
An increase in SVD is known to occur with age and in the presence of cerebrovascular risk factors.8 In non-demented persons, microvascular pathology influences specific cognitive functions such as those involving speed of mental processing.8 Although the substrate for dementia in SVD is still unclear, in vivo studies9, 10 indicate a similar pattern and degree of cortical dysfunction and volume loss in AD and SVD. The functional deficits reported to date in patients with SVD dementia suggest significant cortical and limbic degeneration, similar to AD, although the underlying pathology is predominantly white matter leucoencephalopathy. Remote hippocampal injury has been found in some cases with extensive microvascular disease,11 but has not been systematically studied. Indeed, there have been no studies on the distribution and extent of neuronal loss in patients with SVD. In this study, neuronal loss from the CA1 sector of the hippocampus was quantified in demented patients who did not reach pathological criteria for AD, or any other neurodegenerative disease, but who had significant SVD. The aim of this study was to determine if SVD per se can cause hippocampal degeneration and as such contribute to the dementia syndrome.
MATERIALS AND METHODS
Case selection and characterisation
Cases were collected from both population based research studies on AD and from a tertiary referral centre for neurodegenerative diseases. This study was approved by the ethics review committees of the Central and South Eastern Sydney Area Health Services. All patients were examined by a neurologist or geriatrician. Additional corroborative information for each patient was obtained from an informant interview to ascertain the pattern and type of deficits and to aid in the estimation of disease duration. Fifty four patients who met NINCDS-ADRDA criteria for possible or probable AD12 came to autopsy during the collection period of seven years.
Brains were fixed in neutral buffered formalin for two weeks, and then the weight, volume, and anteroposterior length of the cerebral hemispheres determined.13 The hemispheres were then sectioned at about 3 mm intervals in the coronal plane. The cerebral cortex, hippocampus, brainstem, and cerebellum were sampled from standardised regions of each brain. Sections were stained with haematoxylin and eosin, modified Bielschowsky silver and ubiquitin immunohistochemistry, and examined to perform the classifications outlined below and to exclude pathologies other than AD and SVD.
Each case was given a grading using each of the following classification schema.
(a) CERAD14: normal, possible, probable, or definite AD. Neuritic plaques were evaluated in sections of the temporal neocortex (Brodmann area 20) stained with the modified Bielschowsky silver stain. Ten random microscope fields at 100× magnification graded as having none (≤1/field), rare (2–5/field), mild (6–15/field), moderate (16–50/field), or severe (>50/field) numbers of neuritic plaques.
(b) Braak15, 16: normal (stages 0–2), transitional limbic (stages 3 or 4), or neocortical (stages 5 or 6) AD. Neurofibrillary tangles were evaluated in sections of the hippocampal formation and temporal neocortex stained with the modified Bielschowsky silver stain. Ten random microscope fields at 200× magnification graded into none (nil found), rare (<1/field), mild (1–2/field), moderate (3–10/field) or severe (>10/field) numbers of neurofibrillary tangles.
(c) Esiri4: normal (grades 0 or 1), SVD (grades 2 or 3). Sections from the dorsolateral prefrontal association cortex (Brodmann area 9), anterior cingulate gyrus (Brodmann area 24), and primary motor cortex (Brodmann area 4) were stained with haematoxylin and eosin. Briefly, cases were classified as normal if they exhibited little or no widening of perivascular spaces, thickening of vascular walls, and/or a few perivascular macrophages. Notably, myelin pallor, attenuation of nerve fibres, and gliosis were absent. Cases were classified as SVD when widening of perivascular spaces (fig 1C), thickening of vascular walls (fig 1A), accumulation of perivascular macrophages (fig 1B), and perivascular myelin pallor or attenuation of nerve fibres (fig 1D) were present.

Photomicrographs showing white matter pathology used in the grading of small vessel disease pathology. (A) Haematoxylin and eosin stained section through the frontal white matter showing an arteriole with a thickened vessel wall. Scale equivalent to (B). (B) Haematoxylin and eosin stained section through the frontal white matter showing haemosiderin laden macrophages around a white matter blood vessel. (C) Haematoxylin and eosin stained section through the frontal white matter showing perivascular dilatation with attenuation of surrounding white matter. (D) Modified Bielschowsky silver stained section through the frontal white matter showing attenuation of myelin in the deep white matter.
For this study, case classification for SVD was based on the presence of SVD using the Esiri grading scheme and the absence of probable or definite CERAD AD or a Braak neocortical neuritic stage of AD. Four of the 54 cases fulfilled such criteria for SVD in the absence of any other significant degenerative condition. For comparison, five AD cases were selected on the basis of absence of SVD and the presence of definite CERAD AD and a Braak neocortical stage of AD (table 1). Although 12 of the 54 cases showed both SVD and AD using the classifications outlined above (four with additional large vessel infarction, two with dementia with Lewy bodies,17 and two with hippocampal sclerosis), these cases were not included in the study. This enabled examination of the role of SVD, and its comparison with AD, without the complication of an overlap in the type of cortical pathology in the two dementia groups. Seven controls matched for age and sex from the population based research studies who were without significant neuropathology (normal using all schema) were also selected for comparison.
Screening procedure and classification schema
Quantification of hippocampal formation
The techniques used to quantify hippocampal volume and CA1 neurone number have been described in detail in our previous publications.18, 19 Briefly, the entire hippocampal formation was dissected from the right hemisphere, the blocks cryoprotected in 30% sucrose, and three 48 μm thick sections cut from the caudal face of each block on a freezing microtome. Series of sections from each case were stained with haematoxylin and eosin, cresyl violet, and nickel peroxidase.20 The boundaries of each of the hippocampal subregions were delineated on the cresyl violet stained sections and drawn after magnification on a microfiche reader (×19 magnification). The volume of each region was then determined using point counting,18, 19 and the total number of neurones in the CA1 region estimated using the optical dissector technique.18, 19, 21, 22 The first dissector frame (120 μm × 120 μm) was placed randomly in the CA1 sector, and then systematic samples at 2.4 mm horizontally and 1.2 mm vertically were counted. The coefficient of variance for the control group was 0.208, and the coefficient of error was 0.074.
Group means were compared using analysis of variance, and correlations examined using linear regression analysis (Statview 5, SAS Institute Inc, Cary, North Carolina, USA).
RESULTS
Case descriptions
Table 2 gives the details of the classification for each case. The mean age did not differ between any of the groups (F=2.61; p=0.112), although patients in the SVD group were on average older than those in the AD groups (86 (12) years v 76 (4) years; controls had an mean age of 75 (8) years) (values are mean (SD)).
Age, sex, and neuropathological grading for each case
Summaries of the clinical presentation and progression of each case with SVD are listed below to illustrate more clearly the type of case studied.
S1 was a 69 year old woman who presented with increasing impairment of memory. Her past medical history consisted of type II diabetes, hyperlipidaemia, and hypertension, which was controlled by captopril. After evaluation she was diagnosed with AD. The following year she suffered a stroke-like episode, but computed tomography showed no focal lesion. Cerebral atrophy and leucoaraiosis were described. She continued to decline and was cared for at home by her husband. Three years after presentation she was admitted to a regional hospital after a fall. She died nine days later after a five day history of decline in neurological function. Death was attributed to an acute myocardial infarct. Her neuropathological examination showed a 3 mm lacune in the right putamen and an area of destruction of the white matter at the angle of the right lateral ventricle.
S2 was a university professor who retired at 65 years of age, although had continued to participate in academic activities. He presented at age 75 with a two year history of increasing difficulty with word finding and had ceased lecturing because of this. He had also developed difficulties with navigation while driving. Formal assessment at this time revealed a mini mental state examination23 of 30/35, and a diagnosis of AD was made. He had no history of diabetes or hypertension, although he had suffered a transient ischaemic attack two years earlier. His medications were aspirin, melleril, and voltaren. Over the ensuing three years he deteriorated, became inert, and was unable to handle money, although he was able to dress and groom himself. Neurological examination showed bradykinesia, rigidity, and ataxia. He died at age 80, and neuropathological examination showed small lacunes in the left internal capsule, right claustrum, and right globus pallidus. There was some loss of pigmented neurones from the substantia nigra but no Lewy bodies.
S3 was a 93 year old woman referred for evaluation. She had a 10 year history of a decline in domestic duties and increasing neglect of her self care. She had delusions and wandered if left unattended. On examination, she had deficits in memory, insight, visuospatial abilities, drive, and planning. Her mini mental state examination was 14/30. Her previous history included a myocardial infarct eight years earlier, but was otherwise unremarkable. She had several admissions to hospital after falls, including a fractured pelvis. She was admitted to a nursing home one year before death. Neuropathological examination showed an old cavitated infarct involving the internal capsule, caudate, and putamen, which measured 1.5 cm in anterioposterior extent. Haemosiderin laden macrophages were present at this site.
S4 was a 94 year old woman who presented with a one year history of increasing forgetfulness. She had a sustained delusion that her symptoms were due to her use of a pesticide spray. She also had visual hallucinations. Investigation showed deficits in memory, praxis, and an unsteady gait consistent with having had bilateral knee replacements. She had no history of diabetes, cardiac disease, or hypertension and was taking no medicines. She died from a cardiac arrest while resident in a nursing home. Neuropathological examination showed generalised atrophy, with no focal lesions.
Diagnostic pathology
Figure 1 shows examples of the white matter pathology in the SVD cases. Changes included both those involving the vessels themselves and those involving the surrounding tissue. Thickening of arteriolar walls (fig 1A), accumulation of macrophages (fig 1B), perivascular dilatation (etat criblé, fig 1C), and attenuation of myelin (fig 1D) were seen in all cases. These changes were observed in all of the white matter regions examined.
In the SVD cases, no cortical plaques were detected in two cases and moderate numbers were found in the other two (table 2). Cortical neurofibrillary tangles were absent in three cases and mild in the fourth (table 2). In contrast, the patients with AD had severe cortical neurofibrillary tangles in four of the five cases and moderate in the fifth (table 2). Cortical plaques were severe in two of the five and moderate in the remainder (table 2).
Hippocampal volume changes
The grey matter component of the hippocampal formation is significantly smaller in both of the dementia groups than in the control group (table 3). When the subregion volumes were analysed, all regions except the dentate gyrus and hilus (CA4) were smaller in patients with AD than in controls. In SVD, significant atrophy was confined to the large CA1 and subiculum grey matter regions (table 3). The magnitude of the atrophy in these hippocampal regions was not significantly different between the dementia groups.
Volumes (mm3) of the hippocampal subregions for each group
CA1 neurone number
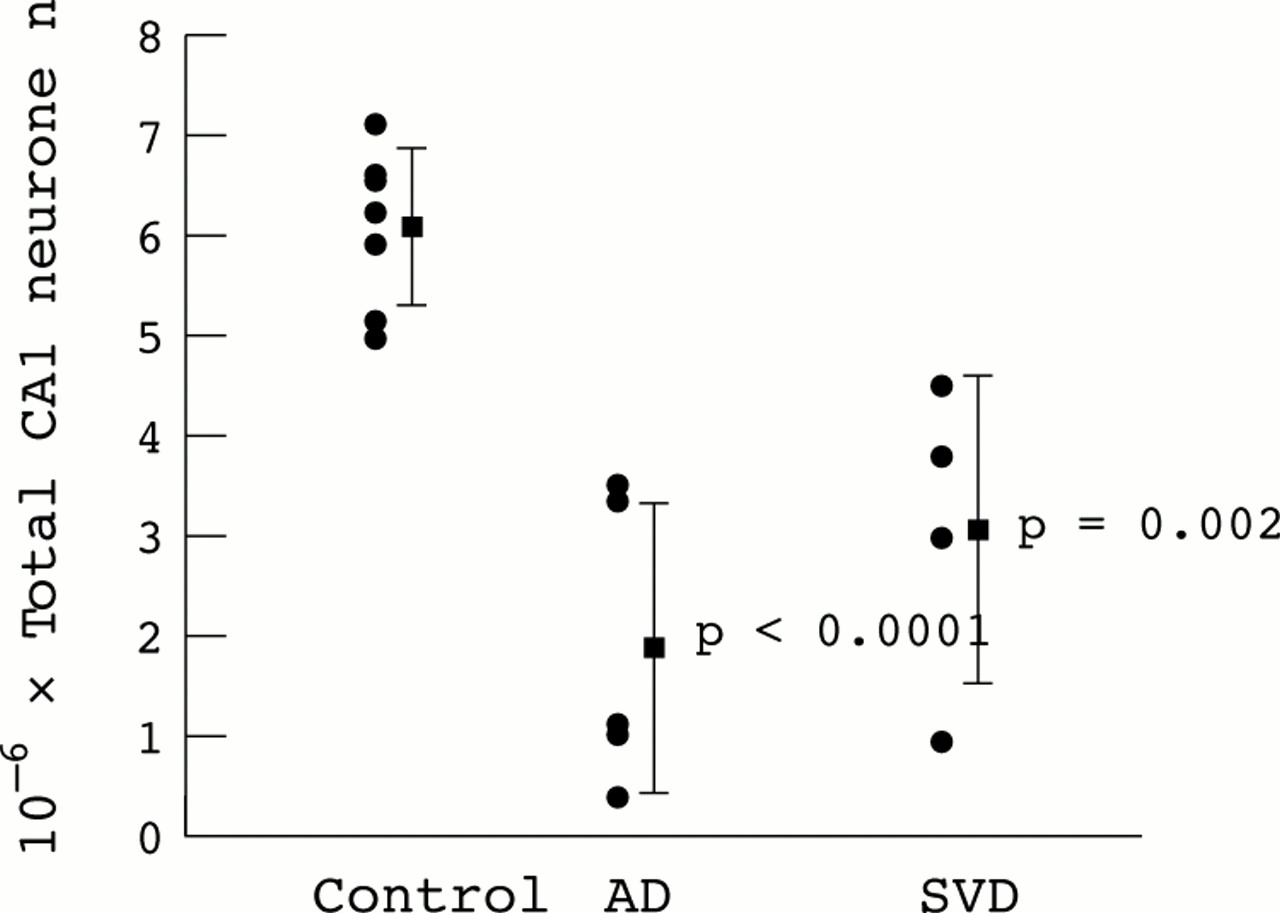
The total estimated neurone number in the CA1 is also significantly reduced in both dementia groups compared with controls (F=17.3; p=0.0002; fig 2). The magnitude of CA1 loss is not significantly different between the SVD and AD groups (50% and 69% respectively). Considerable variation in neurone number exists within the two dementia groups, although this is not as a result of disease duration (r2=0.035; p=0.63) or age at death (r2=0.001; p=0.94)

{kind=link}
{kind=link}
Neurone number in the CA1 region of the hippocampus in control, Alzheimer's disease (AD) and small vessel disease (SVD) groups. Bar = mean ± 1 SD, p values for AD and SVD groups relative to control group.
DISCUSSION
Significant atrophy and neuronal loss from the CA1 region of the hippocampus are one of the pathological hallmarks of AD.22, 24 Interestingly in this study we have shown that a similar pattern and magnitude of hippocampal atrophy and neurone loss also occurs in patients who do not reach pathological criteria for AD but who have SVD. These findings, together with the fact that all nine patients with dementia met clinical criteria for probable AD, suggest that SVD can result in a cortical dementia syndrome similar to AD and that hippocampal neuronal loss contributes to this clinical picture.
Hippocampal damage in AD is considered an early event with substantial pathology and early atrophy preceding symptom onset.25, 26 After these early changes, the widespread deposition of abnormal proteins within the association cortices occurs, with cognitive decline leading to clinical dementia and appreciable neurodegeneration.16, 27, 28 This knowledge of the progression of pathology underlying the clinical dementia and cortical degeneration of AD has been derived from studying a large number of cases. Similar studies have not been performed for SVD dementia because of the lack of consensus on diagnostic requirements for SVD and the relative rareness of cases in which SVD is the only substrate for the dementia. In our prospectively studied series of patients with dementia, and other autopsy series,2, 4 the proportion of cases in which significant microvascular disease is the principal pathology is small (<10% in this study). Several studies have investigated the consequences of coexisting cerebrovascular disease and AD. Both the OPTIMA and Nun studies show that cognitive performance is worse in subjects with early AD and cerebrovascular disease (both large vessel and small vessel disease) than in patients with AD alone,29, 30 suggesting that the vascular pathology contributes to dementia severity. Similarly, in subjects with equivalent cognitive performance, the burden of AD-type pathology is less in those with AD and coexisting cerebrovascular disease than in those with AD alone,7, 31 also supporting the concept that vascular pathology contributes to the dementia. Our results suggest that this contribution is due to pyramidal cell loss through non-AD mechanisms.
The study of vascular dementia has been dominated by large vessel disease (infarction) and only recently has the importance of SVD been realised.4, 11 The loss of neurones with large vessel disease is unquestioned, although neuronal loss due to microvascular pathology has been anecdotal and not systematically studied. Our study not only provides evidence for the existence of SVD as an independent cause of dementia, but also provides some insights into the pathogenesis of this dementia by showing that substantial hippocampal pyramidal cell loss occurs in SVD. This suggests a causative link between SVD, pyramidal cell loss, and hippocampal degeneration, and raises the possibility that cortical microvascular pathology contributes to dementia through pyramidal cell loss. In vivo studies showing a similar pattern and severity of cortical dysfunction and atrophy in AD and SVD support this suggestion.9, 10 Overall, there may be little difference in the degree of neuronal loss between SVD and AD, with only the mechanism of degeneration differing between these dementia syndromes.
The mechanism of neuronal degeneration in SVD may be apoptosis, consistent with an absence of pathological debris. Apoptosis is increased in ischaemia,32 and markers of apoptosis are increased in areas of leucoaraiosis compared with adjacent white matter.33 An increase in cell cycle proteins within hippocampal neurones in patients with SVD31 is also consistent with abnormal cellular processing leading to apoptosis. The location of these changes within hippocampal CA1 neurones suggests they may precede the cell loss shown in this study. It is now important to study large cohorts of patients with SVD in order to determine the temporal sequence of the neuronal damage shown and the onset of dementia. Such knowledge is essential for effective treatment strategies.
Acknowledgments
This work was supported by a programme grant from the Medical Foundation of the University of Sydney and by the National Health and Medical Research Council of Australia. JJK was a Medical Foundation Fellow, and GMH is a NHMRC Principal Research Fellow.