Article Text

Abstract
Objective: To study the clinical features and genetics of idiopathic generalised epilepsy (IGE) beginning in adult life.
Methods: Consecutive patients with IGE, defined as generalised seizures with spike or polyspike and wave on EEG, were studied in the setting of a first seizure clinic where an early postictal EEG record is part of the protocol. Patients were divided into two groups: “classical IGE” with onset before 20 years and inclusive of all the IGE subsyndromes recognised by the international classification; and “adult onset IGE”, when seizure onset was at age 20 years or later. Seizure patterns, clinical features, and genetics of the adult onset group were examined.
Results: Of 121 patients with an electro-clinical diagnosis of IGE, 34 (28%) were diagnosed as adult onset IGE. The seizure patterns in these 34 cases were tonic–clonic seizures + absences (3), tonic–clonic seizures + myoclonus (6), and tonic–clonic seizures alone (25). Tonic–clonic seizures were often precipitated by alcohol or sleep deprivation. The proportion of affected first and second degree relatives did not differ between the classical and adult onset IGE groups. Twenty adult onset cases were treated with sodium valproate, four with other antiepileptic drugs, and 10 were untreated. Follow up of 32 of the 34 cases (for 31 (22) months (mean (SD)) showed that tonic–clonic seizures recurred in eight patients: five with identified provocative factors and three without.
Conclusions: Adult onset IGE is a relatively frequent and benign disorder. Seizures are usually provoked and are easy to control. Patients in this age group may often be misdiagnosed as having non-lesional partial epilepsy. Early postictal EEG and sleep deprivation studies may improve the detection of these patients. Pedigree analysis suggests that adult onset IGE, like classical IGE, has a genetic aetiology.
- idiopathic generalised epilepsy
- adult onset
- genetics
Statistics from Altmetric.com
Idiopathic generalised epilepsy (IGE) is a common form of epilepsy, accounting for 20–40% of all epilepsies.1–,3 IGE is clinically characterised by absence, myoclonic seizures, and tonic–clonic seizures with the electroencephalographic (EEG) pattern of bilateral, synchronous, and symmetrical spike and wave or polyspike and wave discharges. On the basis of the predominant seizure type and age of onset, the international classification recognises four main IGE subsyndromes: childhood absence epilepsy (CAE), juvenile absence epilepsy (JAE), juvenile myoclonic epilepsy (JME), and IGE with tonic–clonic seizures alone (GTCA).4,5 Onset of IGE in childhood or adolescence is characteristic and currently the classification only recognises subsyndromes with onset before adulthood. Onset in adult life is generally considered to be rare—there are only a few reports of such cases.6–,16 Before the development of the current classification, however, Gastaut reported that seizures began after adolescence in 35% of his IGE cases.6
In this paper we describe 34 consecutive patients with IGE beginning at the age of 20 years or after, emphasising their clinical features and genetics. Our experience suggests that adult onset IGE is more common than is generally realised, and that the diagnosis may be overlooked without a directed diagnostic EEG strategy. We also explored the clinical genetics of this group for evidence that adult onset IGE is aetiologically distinct from the classical IGE subsyndromes.
METHODS
We retrospectively reviewed consecutive patients with IGE from the first seizure clinic at the Austin and Repatriation Medical Centre (ARMC), Melbourne. The first seizure clinic recruits patients with new onset seizures from emergency departments of hospitals in the north east sector of Melbourne. We attempt to obtain an EEG within 24 hours, and when the first EEG is negative a sleep deprivation EEG is done.3
Case ascertainment
Patients with IGE were identified through the first seizure clinic database. We included patients with single or multiple generalised seizures, in whom at least one active EEG showing a generalised spike and wave or polyspike and wave pattern was obtained. We reviewed medical records to collect general information such as age, sex, prenatal and perinatal history, and mental and motor development. Particular attention was paid to the collection of the epileptological data, with careful annotation of age of onset and seizure types, EEG results, provocative factors, time of the occurrence of the tonic–clonic seizures, family history, treatment, and outcome. Patients were contacted personally by telephone to update the outcome and clarify the family history. Data were then analysed and patients were divided in two groups by the age of onset of the first seizure. On the basis of published reports1,17 and the concept in the international classification4 that IGE has an “age related” onset, we chose the age of 20 years as a practical division between “classical IGE” (comprising the subsyndromes of childhood absence epilepsy, juvenile absence epilepsy, juvenile myoclonic epilepsy, and IGE with tonic–clonic seizures alone) and epilepsies of later onset, which were designated “adult onset IGE”. The age of onset did not necessarily coincide with the age of presentation at the clinic, as some patients had had diagnosed or previously unrecognised seizures before referral to our clinic.
Characterisation of adult onset IGE
On the basis of seizure type, the patients with adult onset IGE were divided into three groups. Those with absences and tonic–clonic seizures were defined as “adult onset absence epilepsy”; the combination of myoclonic jerks and tonic–clonic seizures was defined as “adult onset myoclonic epilepsy”10; and patients with only tonic–clonic seizures were classified as “adult onset tonic–clonic epilepsy.”
Genetics
We undertook a pedigree analysis of the adult onset IGE and compared it with the classical IGE cohort. Information was obtained, where possible, on the number of first and second degree relatives and the number with a diagnosis of epilepsy. We excluded relatives with single seizures and febrile seizures alone from the analysis.
RESULTS
We identified 121 patients with IGE between March 1994 to December 2000. Their EEGs showed generalised, synchronous, and symmetrical spike and wave or polyspike and wave pattern at a frequency of 2.5 to 5 Hz, with normal background. There were 62 male and 59 female patients, aged from 2 to 75 years. Tonic–clonic seizures led to referral to our clinic in 111 of these 121 patients; eight only had absences, and two presented with myoclonic jerks. The age of onset of seizures in the 121 patients with IGE is shown in fig 1⇓.
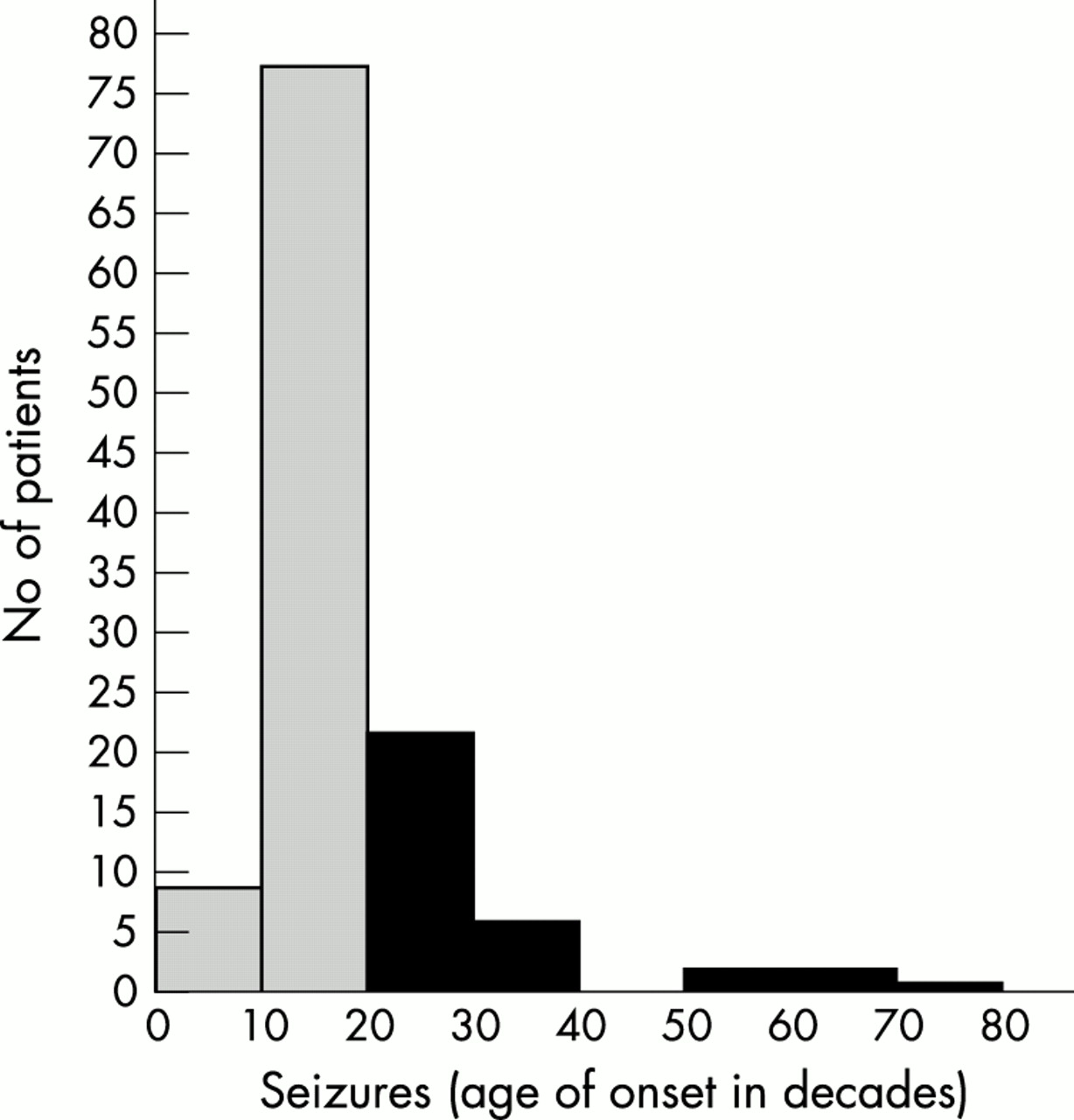
Age of onset of seizures in 121 IGE cases; shaded bars indicates seizure onset before age 20 years (classical IGE) and black bars after age 20 years (adult onset IGE).
IGE with age of onset before 20 years
We identified 87 patients whose age of first seizure was under 20 years (“classical IGE”). There were six patients with childhood absence epilepsy, 16 with juvenile absence epilepsy, 22 with juvenile myoclonic epilepsy, and 42 with IGE with tonic–clonic seizures alone. One case had absence epilepsy with eyelid myoclonia.18,19 Among these 87 patients, seven presented to the clinic after the age of 20. In two cases a relapse of known childhood epilepsy had occurred. The other five patients presented to the clinic after the age of 20, but clinical evaluation revealed that undiagnosed seizures had occurred previously. The delay of the diagnosis for these patients was one to seven years (mean four years). These patients will not be discussed further as their epilepsy syndromes fall within the current classification.
Adult onset IGE
Thirty four of the 121 cases (18 male, 16 female) had seizure onset at age 20 or after. The mean age of presentation to our clinic was 33 years (median 27; range 20 to 75 years). On the basis of a searching clinical history, none of these patients had had seizures before the age of 20 years.
Subsyndrome phenotypes in adult onset IGE
Adult onset absence epilepsy
Three patients had absence seizures beginning after the age of 20 years. The first was a 45 year old woman who was referred to the clinic after her first tonic–clonic seizure. During the first consultation she complained of 10 years of weekly brief blank spells, with losing of track of conversations, followed by immediate recovery. Her EEG showed 3–4 Hz spike and wave discharges. She had a daughter with childhood absence epilepsy, and two sisters and a cousin with seizures (fig 2⇓, family 1). The second was a 20 year old woman who presented after an episode of prolonged absence followed by two tonic–clonic seizures and her EEG showed polyspike and wave discharges. Her maternal grand mother had seizures. The third was a woman presented at the age of 29 while pregnant with absences and her EEG showed generalised spike and wave discharges. A cousin had epilepsy.

Family pedigrees.
Adult onset myoclonic epilepsy
Six patients presented with myoclonic jerks and tonic–clonic seizures, often precipitated by sleep deprivation and alcohol. The mean age of onset was 37 years (median 30 years, range 20 to 75). Myoclonic jerks affected only the arms in all six cases, were infrequent, and predominantly occurred in the morning. The number of tonic–clonic seizures in each case was small: three patients had only had one seizure, two had had two seizures, and the final patient had had three. Neurological examination and neuroimaging (three patients had magnetic resonance imaging (MRI) and three computed tomography (CT)) were normal; in particular, none had tremor or evidence of dementia. The EEG in all six patients showed spike and wave or polyspike and wave discharges. The family history was positive for epilepsy in four of the six cases.
Adult onset myoclonic epilepsy, case 1
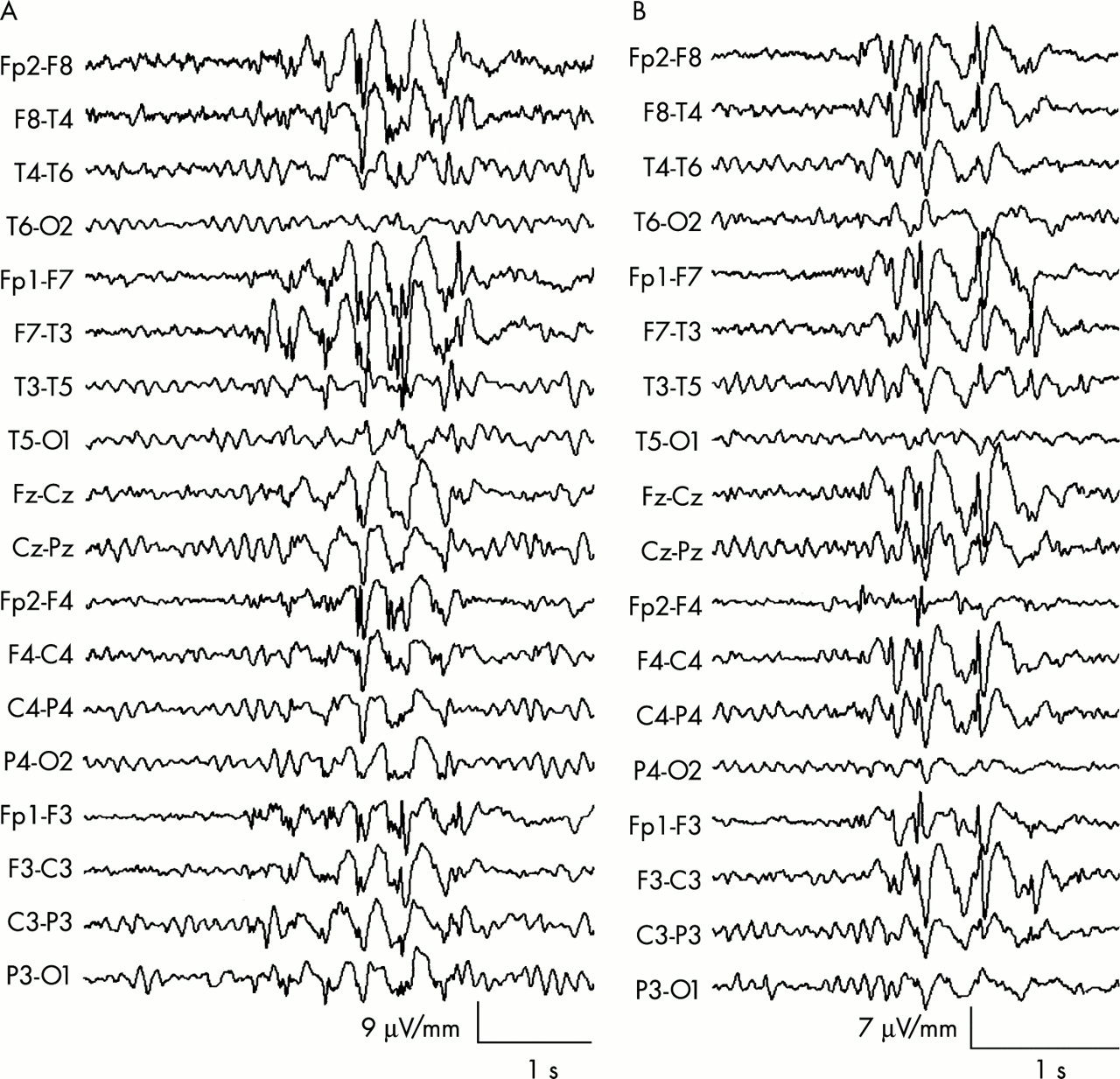
A 75 year old woman was referred to the clinic after recurrent myoclonic jerks affecting the upper limbs with dropping of objects from her hands, followed by a tonic–clonic seizures. Previously she had been well. There was no evidence of intercurrent neurological or other medical illness, and no evidence of misuse of prescribed or other drugs, or of any psychological precipitants. Her EEG showed typical generalised 3–5 Hz spike and wave and polyspike and wave discharges with bifrontal predominance (fig 3A⇓). Her mother had seizures and her parents were first cousins. She became seizure-free on sodium valproate.

{kind=link}
{kind=link}
{kind=link}
(A) EEG of a 75 year old woman with adult onset myoclonic epilepsy (see text, case 1), showing generalised spike and wave and polyspike and wave discharges with bifrontal predominance. (B) EEG of a 69 year old man with adult onset tonic–clonic epilepsy alone (see text, case 1) showing generalised spike and wave discharge with bifrontal predominance.
Adult onset myoclonic epilepsy, case 2
A 50 year old man had three episodes of clusters of jerks in the arms followed by tonic–clonic seizures from age 50 to 59 years. His last cluster of myoclonic jerks happened a day after hand surgery, when the synthetic opioid pethidine (100 mg injection) was used as premedication. His EEG also showed frequent generalised spike and wave activity. Jerks of the arms were recorded and found to be associated with polyspike and wave discharges. He was treated with clonazepam for two weeks during the last cluster. After 18 months of follow up he remained seizure-free.
Adult onset tonic–clonic epilepsy
Twenty five patients had tonic–clonic seizures as the only manifestation of IGE. The mean age at the first seizure was 30 years (median 26 years, range 21 to 69). Thirteen patients had only had one seizure, eight had had two, two had had three, and two had had four. Provocative factors were sleep deprivation with or without alcohol in 15, and photic stimulation in four (two of these also had precipitation by sleep deprivation). Eight patients had no clear provocative factors. Tonic–clonic seizures occurred in the morning within an hour of waking in only four cases. MRI was performed in 15 patients and CT in nine; all scans were normal.
Adult onset tonic–clonic epilepsy, case 1
A 69 year old man presented at the clinic following a tonic–clonic seizure occurring in the morning and not preceded by an aura. There were no provocative factors and no history of similar episodes or minor events. An EEG performed within 24 hours showed 4–5 Hz generalised spike and wave and polyspike and wave discharges with bifrontal predominance (fig 3B⇑). His family history revealed a twin sister and a distant cousin with epilepsy (fig 2⇑, family 2). He was started on sodium valproate and remained seizure-free after 12 months of follow up.
Pedigree analysis of 121 IGE cases
A positive family history for epilepsy, excluding febrile seizures, was obtained from 34 of 87 cases with classical IGE (39%) and from 19 of 34 cases with adult onset IGE (56%). Of the 53 multiplex families, there were 35 with two members affected and 17 with three to six members affected.
We obtained specific information on the proportion of affected first degree relatives in 84 of the 87 cases of classical IGE and 32 of the 34 cases of adult onset IGE; the missing data were the result of adoption or loss to follow up. Similarly, complete data for second degree relatives were obtained from 82 of 87 and 31 of 34 cases. Table 1⇓ shows that there was no significant difference in the occurrence of epilepsy in relatives of classical IGE versus adult onset IGE probands in this cohort. A subanalysis of the family history of IGE patients was done in which late onset IGE was defined as IGE starting at 25 years or later. This comprised 20 cases. The overall proportion of affected relatives in these 20 late onset IGE patients (12/318; 4%) did not differ from the 101 classical IGE group (40/1171; 3.4%).
Epilepsy in first and second degree relatives of idiopathic generalised epilepsy probands
Treatment and follow up in the adult onset IGE
The follow up of 32 of the 34 adult onset IGE cases was between three and 86 months (mean (SD), 31 (22) months; median 25 months). Two patients were lost to follow up. Of these 32 cases, 24 were treated with antiepileptic drugs and eight chose not to be started on drug treatment. Sodium valproate was used in 20 patients and single cases were treated with phenytoin, carbamazepine, lamotrigine, and topiramate. Five patients had recurrent tonic–clonic seizures; provoking factors were alcohol and sleep deprivation in three and withdrawal of drug treatment in two. Only three patients had recurrent seizures on medication, without identified provoking factors. Follow up of the untreated eight patients was from 17 to 76 months (mean 41). None had further seizures.
DISCUSSION
Research on IGE has almost exclusively focused on the childhood and adolescent onset subsyndromes. Adults with isolated or rare tonic–clonic seizures and negative EEG studies are generally regarded as having partial epilepsy of uncertain cause, and adult onset IGE would rarely be considered. Adult patients with an electro-clinical picture compatible with IGE often represent relapse of childhood epilepsy; however, de novo IGE in adult life has been reported by several investigators.6–,16 Attempts to characterise the subsyndromes and genetics of adult onset cases have been sparse.7,10,16
In our first seizure clinic—which serves a predominantly late childhood to adult population—28% of our cases of IGE began at age 20 years or later. This is consistent with Gastaut’s report in 1981 that 35% of his IGE cases began after age 18 years, and of these 95% began before they were 50.6 The relatively frequent identification of adult onset IGE in our clinic probably relates to our practice of doing an early postictal EEG followed by a sleep deprivation EEG in negative cases. This leads to a higher diagnostic yield in first seizure cases.3 The age distribution of seizure onset of our cases in fig 1⇑ should not be interpreted as the true population frequency distribution, as our clinic is biased towards adolescents and adults and so cases of IGE with childhood onset (for example, childhood absence epilepsy) are underrepresented.
There is no clear age boundary where classical IGE ends and late onset IGE begins. The international classification does not define age of onset limits in IGE subsyndromes. Although childhood or “juvenile” onset is stressed, Janz has indicated diagrammatically that he accepts cases in their early twenties as part of classical IGE.17 A similar problem is seen in middle childhood, with the distinction between childhood absence epilepsy and juvenile absence epilepsy; the former typically begins at four to eight years with frequent (“pyknoleptic”) absences, and the latter at 10 to 17 years with rare absences. Children with frequent absences beginning at 11 years or rare attacks at age 9 make a crisp age distinction between childhood and juvenile absence epilepsy impossible.1,17 We chose the age of 20 years as a pragmatic cut off year for “adult” IGE. Fourteen cases began in the 20 to 25 year epoch, and these young adult cases may include part of the “tail” of distribution of classical adolescent IGE. A recent paper on late onset IGE described patients with age of onset ranging from 18 to 50 years but with a median of 20 years, indicating that half their sample was in the 18 to 20 year epoch.16 That our age choice was reasonable is suggested by comparison of our genetic analysis with and without the early 20s epoch, where no difference was seen (table 1⇑). When the genetic architecture and actual genes determining particular syndromes in childhood, adolescence, and adulthood are known, a better classification of IGE may emerge.
Although the lower age limit of adult IGE can be debated, the age of onset distribution is extensive, with our oldest case being 75 years at onset. Loiseau and colleagues searched for cases of IGE beginning after the age of 60 years and found none in their material, but identified five cases in the European literature.9 Five of our 34 cases had an age of onset after 40 years; three had adult onset tonic–clonic seizures and two had adult onset myoclonic epilepsy. Seizures were rare in all five cases and were provoked in three. Family history was positive for epilepsy in three of the five; the frequency of affected first and second degree relatives did not differ in this small sample of five compared with the whole late onset IGE group (data not shown).
Classical IGE subsyndromes are classified according to seizure type. Similarly in our adult onset IGE cases we were able to distinguish different clinical subtypes. The majority of our patients (74%) had tonic–clonic seizures as the only manifestation of their epilepsy. In Gastaut’s series, 98% had nocturnal “grand mal” or tonic–clonic seizures alone, while absences or myoclonic jerks were exceptional.6 In contrast we were able to identify six cases (18%) with myoclonic jerks and tonic–clonic seizures. Gilliam and coworkers described 11 adults with new onset epilepsy with myoclonic jerks, tonic–clonic seizures, and spike and wave or polyspike and wave on EEG, and estimated that those cases comprised 0.5% of all new epilepsy referrals and 10% of all new IGE referrals.10
Only three of our 34 cases had absence seizures. Absence status arising de novo in adult life, particularly in middle aged women, has been described.12–,14 These patients usually present with a confused state and eyelid fluttering or myoclonus of varying degrees of severity. Panayiotopoulos et al also described cases of adult onset IGE with infrequent tonic–clonic seizures, mild and often unrecognised “phantom absences,” and frequent absence status.15
Classical IGE has a genetic aetiology with complex inheritance. Family and twin studies suggest a shared genetic origin for the IGE subsyndromes, with some genetic specificity determining these subsyndromes.1,20 The pedigree analysis of our adult onset IGE cases showed that 19 of the 34 cases (56%) had near relatives with epilepsy. A positive family history of epilepsy in relatives of adult onset IGE patients was found in 28% of the cases reported by Oller-Daurella and Sorel,7 and in some cases reported by other investigators.10,11,16,21
The lack of difference in the frequency of affected relatives between classical and adult onset IGE in our study (table 1⇑) is consistent with a late onset IGE also having a genetic aetiology. Indeed, most of the affected relatives of our late onset cases had seizure onset during childhood or adolescence (data not shown), and although specific epilepsy syndromes were not determined in most cases, some relatives had classical IGE. An example is shown in fig 2⇑, where classical childhood absence epilepsy and adult onset absence epilepsy are found in the same family. This suggests that classical and late onset IGE cases share genetic determinants, but formal testing of this hypothesis would require more detailed phenotyping in a large pool of relatives with both types of IGE.
An autosomal dominant primary generalised epilepsy with adult onset—familial adult myoclonic epilepsy (FAME)—has been described in the Japanese.22,23 FAME is characterised by myoclonus in the upper and lower limbs, rare tonic–clonic seizures, and a linkage to chromosome 8q24.22,23 There is a general agreement that this is a distinct syndrome with characteristic and distinguishing tremulous movement of the fingers and progressively worsening myoclonus—features that are not seen in classical IGE or in patients with adult onset myoclonic epilepsy. The clinical syndrome and the mode of inheritance distinguish FAME from adult onset IGE.
As in previously published cases we found adult onset IGE to be a benign epilepsy with rare seizures, usually triggered by sleep deprivation or alcohol excess.6,7 We identified a variety of subsyndromes based on the predominant seizure type, with adult onset tonic–clonic seizures being the most common. Seizures were controlled on drug treatment or by improving the life style, and after 31 months of mean follow up only 10% had occasional seizures without provocation. Our data suggest that adult onset IGE is a genetic disorder with infrequent seizures which, when they occur, often result from provocation. An obvious question is why seizures did not occur in the teenage years, when provoking factors are usually frequent. Perhaps the oligogenic architecture of IGE includes genes with maximum expression in adult life, and these are the ones particularly affected in adult onset cases.
The recognition of adult onset IGE has its implication for accurate diagnosis and treatment. Adults with single or widely spaced tonic–clonic seizures and a family history of epilepsy are prime candidates for adult onset IGE. When an adult has new onset seizures, tumours and other space occupying lesions or degenerative disorders are major diagnostic considerations. The diagnosis of adult onset IGE from electro-clinical criteria can alleviate concern about such disorders, avoid unnecessary investigation, direct appropriate treatment, and allow an optimistic prognosis. Moreover, our pedigree analysis suggests that adult onset IGE, like classical IGE, has a genetic origin and such cases should be included in analyses where genes for IGE are being sought.
Acknowledgments
We thank the EEG and MRI technicians of the Austin and Repatriation Medical Centre (ARMC), the emergency room staff at the ARMC, Box Hill, and Maroondah Hospitals, all referring physicians, and Regula Briellmann for information on relatives of some IGE patients. Novo Nordisk Pharmaceuticals and Sanofi Synthelabo provided unrestricted grant support to the first seizure clinic. The study was supported by the NHMRC, Bionomics Ltd, and a University of Melbourne Scholarship (to CM).
REFERENCES
Footnotes
-
See Editorial Commentary p 147
-
Competing interests: none declared