Article Text

Abstract
Background Heat shock protein 27 (HSP27) mutations have been reported to cause both Charcot-Marie-Tooth disease (CMT) type 2F and distal hereditary motor neuropathy (dHMN) although never previously in a single family.
Objective To analyse clinical and electrophysiological findings obtained in a single large Sardinian family bearing the HSP27 R127W mutation.
Methods Twenty-one members of a five generation Sardinian family have been studied, including thirteen members affected by peroneal muscular atrophy and proved heterozygous for the known HSP27 R127W mutation. Twelve patients and eight unaffected relatives were subjected to clinical examination. A standardised electrophysiological study was performed in eleven patients and six unaffected relatives.
Results Mean age at onset (±SD) was 31.2±7.2 years. Mean age at investigation was 45.2±12.9 years and mean disease duration at the time of investigation was 14±12.9 years. According to current criteria for CMT2 and dHMN, of the 10 patients who had undergone both clinical and neurophysiological examination, five were diagnosed as CMT2, two as dHMN and a further two patients were labelled as an intermediate type. Finally, due to the presence of spastic paraplegia, the index patient did not meet established criteria for the diagnosis of CMT or dHMN.
Discussion Findings obtained in the present study, broadening the spectrum of clinical manifestations of disorders associated with HSP27 mutations, support the hypothesis of a continuum between CMT2 and dHMN forms and suggest a possible common spectrum between these entities and several forms of CMT plus pyramidal features (HMSN V), providing important implications for molecular genetic testing.
- genetics
- hmsn (charcot-marie-tooth)
- neuropathy
- neurophysiol, clinical
Statistics from Altmetric.com
Introduction
Heat shock protein 27 (HSP27), also known as HSPB1, belongs to the superfamily of small heat shock proteins and is mainly implicated in the assembly of neurofilament network.1 HSP27 mutations have been reported to cause both Charcot–Marie–Tooth disease (CMT) type 2F and distal hereditary motor neuropathy (dHMN)1–6 although never previously in a single family. The aim of this study was to divulge clinical and electrophysiological findings obtained in a single large Sardinian family bearing the HSP27 R127W mutation, revealing how affected subjects featured a marked interfamilial phenotypic heterogeneity varying from pure axonal CMT2F or dHMN phenotype, with one single patient clearly presenting associated features of spastic paraplegia.
Patients and methods
The index case, subject IV-2, a 44-year-old man, was investigated initially and his family tree is shown in figure 1, clearly illustrating how other relatives were affected, leaning considerably in favour of an autosomal dominant means of inheritance. Divergent clinical features were observed in other affected family members. In spite of the index case featuring a marked association of spastic gait with peripheral neuropathy, pyramidal signs were virtually absent among the relatives, with predominant distal muscle weakness and wasting of the lower limbs, compatible with a diagnosis of peroneal muscular atrophy. Over the past 3 years, 21 members of this five generation Sardinian family have been studied, 20 of whom, including 13 members affected by peroneal muscular atrophy syndrome, have undergone screening for CMT2 mutations. Informed written consent was obtained from all participants.

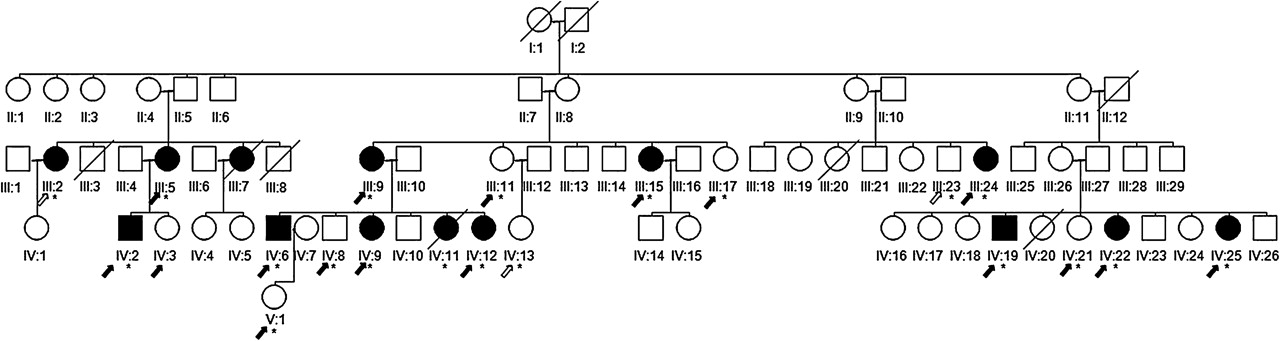{kind=link}
Pedigree of the family with heat shock protein 27 (HSP27) p. R127W mutation. Black symbols indicate affected family members and symbols with a strike through indicate those deceased. Circle symbols, women; square symbols, males. *Genotyped individual. Black arrows, subjects who underwent clinical and electrophysiological studies; white arrows, subjects studied clinically. Index patient is subject IV:2.
Clinical and electrophysiological examination
Data collected included onset of symptoms, duration of disease, initial symptoms, disease course, presence of muscle cramps or pain, impairments (ie, muscle weakness and sensory dysfunction) and other additional features. Twelve patients and eight unaffected relatives were subjected to clinical examination and examined for: weakness and atrophy of the distal and/or proximal parts of the lower and upper limb muscles, deep tendon reflexes of the lower and upper limb muscles, presence of extensor plantar responses and Romberg sign, presence of foot and hand deformities, nerve hypertrophy and scoliosis. Vibration, touch, joint position and pinprick sensation were examined at various sites in the lower and upper limbs. In addition, the presence of tremor, trophic alterations of the feet and knees, cranial nerve abnormalities, involvement of vocal cords, ataxia, contractures, deafness, optic nerve atrophy, pyramidal signs, dysautonomia and dementia was assessed. To determine disease severity in terms of ability to walk and run, each patient was assessed by means of the Functional Disability Scale.7 A similar scale for the evaluation of a personal neurological disability affecting the upper limbs, with a 6 point scale ranging from 0 to 5, was assessed and determined as follows: 0=normal; 1=normal but with cramps; 2=fatigability in writing; 3=handling difficult; 4=need help to use hands; 5=completely dependent. A standardised electrophysiological study was performed in 11 patients and six unaffected relatives. Motor nerve conduction velocities and compound muscle action potentials were investigated in the median and peroneal nerves. Antidromic sensory nerve conduction velocity and sensory nerve action potentials (SNAPs) were investigated in the median and sural nerves. A concentric needle electromyography was performed using standard techniques. Data correlation tests were performed using Pearson's correlation coefficient or Spearman's rank correlations. Diagnoses of CMT and dHMN were made according to common diagnostic criteria.8
Molecular genetic analysis
Genomic DNA was extracted from peripheral blood by standard methods. A panel of 15 highly polymorphic short tandem repeats was used to exclude duplication or deletion of chromosome 17p11.2–p12.9 The coding exons of mitofusin 2 (MFN2), myelin protein zero, gap junction protein beta 1, neurofilament light polypeptide and ganglioside induced differentiation associated protein 1 genes were analysed by direct sequencing (details of primer sequences used to amplify the MFN2, myelin protein zero, gap junction protein beta 1, neurofilament light polypeptide and ganglioside induced differentiation associated protein 1 genes are available on request). In addition, only exon 3 of the Berardinelli–Seip congenital lypodystrophy-2 gene was examined for the known Silver disease mutations10 (conditions available on request). Finally, to analyse HSPB1 coding sequence, exons 1, 2 and 3 were amplified using primers designed on flanking intronic sequences as follows: exon 1: forward ACG GGT CAT TGC CAT TAA TAG AGA CC and reverse AGA AGC GAC CCG CAC TCC CAA TTC; exon 2: forward TGT TAA TCC CTA CCA GCC TGC AG and reverse, AGG CAA GCG TTA CAT TAC ACA CCG; exon 3: forward ACG CGG AAA TAC ACG TGA GTC CTG G and reverse TGC CTG AGG CTT CCT TCC ACA AAC ACC. PCR and sequencing reactions were performed under standard conditions. PCR purification was carried out using ExoSAP-IT (Amersham, Little Chalfont, Buckinghamshire, UK) whereas sequencing reactions were performed using the BigDye v1.1 Terminator Cycle Sequencing Kit (Applera, Foster City, CA, USA). Sequences were loaded onto a 3130XL Genetic Analyser and analysed using the Sequencing Analysis Software V.5.2.
Results
Among the 20 subjects of the family screened for CMT2 mutations, all 13 members considered as being clinically affected by peroneal muscular atrophy syndrome proved heterozygous for the known HSP27 p. R127W mutation in exon 2. Neither of the other genes studied in these subjects revealed the presence of pathogenic mutations or polymorphisms. Furthermore, none of the seven remaining unaffected relatives in our series harboured a mutation in exon 2 of HSP 27 gene. Clinical features of the twelve patients are listed in table 1.
Clinical features of patients with HSP27 R127W mutation
Age at onset was clearly available for 10 of the patients examined. Mean age at onset (±SD) was 31.2±7.2 years. Mean age at investigation was 45.2±12.9 years and mean disease duration at the time of investigation was 14±12.9 years. The course of the disease was slowly progressive in all patients. In five patients aged 30–54 years, sensory examination was entirely normal while a further two other patients featured a very mild vibration sensory loss in the feet. In the remaining five patients with sensory impairment, examination revealed a predominant loss of sensation in the legs while in the arms mild subjective hypopallesthesia was present in one patient. One patient alone presented total loss of sensation in the lower limbs. Clinical characteristics and sensory impairments were more pronounced with age. Involvement of the vocal cords, contractures, deafness, optic nerve atrophy and dementia were not observed in any patients. With the exception of the index patient, no subjects presented bladder or sexual dysfunctions. Mean Functional Disability Scale (±SD) score was 3±1.8 while mean scores for neurological disability in the upper limbs was 1.8±1.7.
Electrophysiological studies (table 2) revealed a marked predominance of motor and sensory abnormalities in the legs. Peroneal compound muscle action potential amplitudes were generally reduced and were totally absent in four patients. Evaluation disclosed normal motor conduction studies of the median nerve in almost all patients, recording motor nerve conduction velocities of less than 38 m/s in only one patient, likely due (as the reduction in peroneal conduction velocity under 38 m/s in patient III5) to axonal loss with secondary demyelinating process. Sural SNAPs were absent in only two patients although their amplitude was markedly reduced in a further three patients. Three of the remaining affected individuals displayed sural SNAPs within the normal range although closer to lower reference limits. Median nerve SNAPs were normal except for a slightly reduced amplitude in two patients. Statistical analysis showed that duration of disease correlates with both sural SNAP amplitudes (r=−0.903; p<0.001) and median SNAP amplitudes (r=−0.669; p<0.05).
Electrophysiological findings of Sardinian patients with the HSP27 mutation
According to the Classification and Diagnostic Guidelines for CMT2 and dHMN,8 of the 10 patients who had undergone both clinical and neurophysiological examination, five were diagnosed as CMT2, two as dHMN (III:24 and IV:12) and a further two patients (IV:25 and IV:9) were labelled as an intermediate type. In fact, in the latter two patients, clinical/electrophysiological findings (mild vibration sense loss in the feet but normal sensory nerve potentials at electrophysiological studies) were insufficient to warrant their clear inclusion in one of the above categories, owing to the fact that current classification requires the absence or presence of small sensory nerve action potentials as inclusion criteria for CMT2, but at the same time formulates the detection of impaired sensation as exclusion criteria for dHMN.
Finally, due to the presence of spastic paraplegia, the index patient did not meet established criteria8 for the diagnosis of CMT or dHMN. Indeed, these criteria8 exclude patients with significant involvement of the CNS, including the pyramidal tract. In this patient, other differential diagnoses such as spinal cord compression, HTLV-1 associated myelopathy or other disorders were excluded.
Discussion
We have described a large Sardinian family with predominant autosomal dominant axonal neuropathy in which a missense mutation (R127W) in the HSP20-α-crystallin domain of the gene encoding HSP27 was identified. The study of this large family allowed us to delineate the clinical spectrum, natural history and electrophysiological findings of this HSP27 mutation. To our knowledge, this is the first described family with a HSP27 mutation where axonal CMT and dHMN were both present in different family members with a significant correlation between duration of disease and the occurrence of reduction of SNAP amplitudes, implying that sensory impairments may not be present in the initial stages of the disease, rather being manifested at a later stage. This point is of particular importance, demonstrating a difference in intensity rather than in nature between CMT2 and dHMN in the cases examined. Moreover, at least two members of the family studied here, diagnosed as affected by clearcut CMT2 forms, had previously been labelled as dHMN in the past, as provided for by standard criteria.8
Furthermore, in several members of the large Sardinian family described in this paper, the clinical phenotype seemed to produce peculiar features not fully corresponding to either of the two forms, raising important questions concerning the specificity of criteria8 that differentiate CMT from dHMN forms. In this regard, the exclusively motor involvement in dHMN is still a matter of debate due to minor sensory abnormalities being described elsewhere.11 Compared with the only article2 reporting the clinical features of seven Chinese patients with HSP27 R127W mutation, the present family was characterised by an earlier age at onset. In addition, while mild to moderate sensory impairment of the hands was observed in all Chinese patients, this observation was not replicated in our study. Indeed, with one exception, none of our patients exhibited sensory abnormalities in the upper limbs.
Bearing in mind the correlation between duration of disease and reduction in SNAP amplitudes both in the sural and median nerves, the latter findings can be easily explained by an older age of Asian patients at examination (63.9±10.7) compared to that of the Sardinian subjects (45.2±12.9). Mutations in another HSP (HSP22)12 13 may present both with CMT2 and dHMN phenotypes; likewise, mutations in glycil-tRNA synthetase gene have been reported to be associated with both diseases.14 15 Mechanisms whereby the same gene mutation is capable of causing different diseases remain unclear. On the grounds of the hypothesis that the primary pathological process resides respectively only in the axons or in the neuron cell body, CMT2 types are usually classified as axonal neuropathies (axonopathies) while dHMNs are considered as a separate entity and classified as motor neuronopathies although considerable variations may occur.12 However, as our knowledge on gene mutations increases, growing evidence is accumulating to support the notion whereby a similar distinction between axonal CMT and dHMN is somewhat dogmatic.
Our findings are suggestive of the existence of a continuum between the two entities, thus advocating a reconsideration of this strict pathonosological classification. The latter concept of a common spectrum is in full accordance with data reported by Irobi,11 underlining the arbitrary nature of the previously mentioned pathological dichotomisation due to the fact that the clinical features manifested in dHMN are characterised by a well established distal distribution indistinguishable from CMT2. Irobi not only suggests a major involvement of axonal sites in pathological damage but moreover observes how, owing to lack of autopsy studies, little is known about the status of the anterior horn motor neuron bodies.11 An additional element of discussion is represented by the occurrence of spastic paraplegia in the index patient carrying the same HSP mutation. Although manifested infrequently, the presence of pyramidal features has been described in several patients with an axonal form of CMT with variable severity ranging from hyperreflexia to overt spastic paraplegia.
Dyck and Lambert16 classified families affected by CMT with associated spastic paraplegia as hereditary motor and sensory neuropathy type V, while CMT patients with mild pyramidal signs but without spastic paraplegia were labelled as peroneal muscular atrophy with pyramidal features by Harding and Thomas.17 A systematic revision of case histories18 has led to postulation that the presence of brisk reflexes in the legs and extensor plantar responses may be viewed as forming part of the CMT2 phenotype. The latter opinion would seem to be in agreement with findings obtained in the present study in which a patient with brisk knee reflexes was indeed observed.
Recent observations indicate that CMT with pyramidal signs is genetically heterogeneous19 and may be caused by mutations of at least two different genes: MFN219 and N88S Berardinelli–Seip congenital lypodystrophy-2 mutation.20 Thus although an isolated occurrence, the certain onset of spastic paraplegia associated with the CMT form in the patient examined would seem to convey an additional feature of phenotypic variability in subjects with HSP27 mutations. However, the authors of the present study were not in a conclusive position to speculate whether this occurrence was specifically linked to the single mutation rather than to other epigenetic factors. Further studies should be performed with the aim of better defining this intriguing observation.
In conclusion, the findings obtained in the present study, broadening the spectrum of clinical manifestations of disorders associated with HSP27 mutations, support not only the hypothesis of a continuum between CMT2 and dHMN forms, but appear moreover to suggest a possible common spectrum between these entities and several forms of CMT plus pyramidal features (HMSN V), providing important implications for molecular genetic testing. Taken together, the data obtained suggest the need to provide for the establishment of more accurate and widely accepted working definitions to be applied in the case of CMT2/dHMN and the other CMT plus forms. These definitions should be based largely on clinical information and integrated with genetic data in order to further enhance the understanding of the pathological mechanisms involved and to provide for the development of targeted therapeutic interventions in the future.
References
Footnotes
Competing interests None.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the university ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.