Article Text

Abstract
Background Acute motor axonal neuropathy (AMAN) and acute motor and sensory axonal neuropathy (AMSAN) are due to an antiganglioside antibody mediated attack, thought to be restricted to motor fibres in AMAN. Sensory symptoms and minor sensory conduction abnormalities, however, have been reported in some AMAN patients.
Objective To verify whether sensory fibres are truly spared in AMAN and whether AMAN and AMSAN represent a continuum.
Methods Serial conduction studies in 13 AMAN and three AMSAN patients were reviewed. To evaluate the variation in sensory nerve action potential (SNAP) amplitude in serial recordings, the least significant change in a test–retest study of 20 controls was calculated. Least significant change for median, ulnar and sural nerves were 44%, 47% and 58%, respectively.
Results In 34% of initially normal sensory nerves of six AMAN patients, SNAP amplitude significantly increased by 57–518%. In three nerves of three AMAN patients, SNAP significantly decreased by 50–69%. Overall, serial recordings allowed detection of sensory fibre involvement in 49% of nerves and in 69% of AMAN patients. In one AMSAN patient, SNAP increased in two nerves by 150–300%; in another patient, SNAPs, unrecordable at baseline in six nerves, reappeared during follow-up and normalised in three nerves. In five nerves of three AMAN and in eight nerves of two AMSAN patients, SNAP amplitudes increased rapidly, suggesting reversible conduction failure of sensory fibres. In other nerves, SNAP increased over months, as for axonal regeneration.
Conclusions Sensory fibres are often involved subclinically in AMAN. Reversible conduction failure may develop in sensory as well as motor fibres in both AMAN and AMSAN. AMAN and AMSAN represent a continuum in axonal GBS.
Statistics from Altmetric.com
Introduction
Guillain–Barré syndrome (GBS) has been divided into several subtypes according to clinical, electrophysiological and pathological findings, infective antecedent and presence of specific antibodies.1 2 Two primary axonal subtypes have been described: acute motor axonal neuropathy (AMAN) and acute motor and sensory axonal neuropathy (AMSAN).3–7 AMAN and AMSAN have been associated with antecedent Campylobacter jejuni infection and autoantibodies to gangliosides, especially to GM1 and GD1a.4–9 AMAN is clinically characterised by exclusive motor involvement although sensory symptoms have been also reported.3 4 10 11 Immunopathological studies in humans and in the rabbit experimental model indicated that AMAN is due to a complement mediated attack of antiganglioside antibodies to the axolemma of nodes of Ranvier of motor fibres with complement deposition, sodium channel cluster disruption, nodal lengthening, macrophage recruitment and final axonal degeneration in motor fibres.12–15 The electrophysiological characteristics of AMAN are absence of demyelinating features and decreased distal compound muscle action potentials (dCMAP).3 4 However, reduced dCMAP amplitudes, conduction block (CB) at common entrapment sites or isolated absence of F waves which promptly recover without the development of temporal dispersion or other demyelinating features, have been described in some AMAN patients with antiganglioside antibodies and ascribed to an antibody mediated reversible conduction impairment at the axolemma of nodes of Ranvier.16–18 AMSAN is quite rare (1–4% of GBS in Japan) even though it has been reported in up to 11% of cases in Bangladesh.9 19 20 AMSAN is clinically characterised by weakness, areflexia and sensory loss. Most of the reported patients had severe weakness requiring mechanical ventilation, inexcitable motor and sensory nerves and poor outcome.21–23 In AMSAN, as in AMAN, the pathology is consistent with an antibody mediated primary axonal damage at the node of Ranvier with the difference that the dorsal as well as the ventral roots are affected.22 23 However, minor conduction abnormalities in sensory nerves have been reported in 5–12% of AMAN patients, blurring the dichotomy between AMSAN and AMAN.3 4 11
The aim of this study was to investigate whether sensory fibres are completely or relatively spared in AMAN by comparing results of serial recordings, and to verify whether axonal GBS subtypes form a continuous spectrum.
Subjects and methods
Patients
We reviewed the clinical and electrophysiological records of patients diagnosed with GBS at the University Hospital of Chieti between January 1995 and June 2009. Patients were classified as AMAN on the basis of the following criteria: (1) acute onset of progressive motor weakness of more than one limb; (2) normal sensory examination; (3) dCMAP amplitudes <80% of the lower limit of normal (LLN) in at least two nerves without evidence of demyelination (AMAN with axonal degeneration) or evidence of reversible conduction failure characterised by reduced dCMAP amplitude with normal or slightly prolonged distal motor latencies (DMLs) and/or partial CB (defined as an amplitude ratio of CMAPs from proximal and distal stimulation less than 0.5) in intermediate nerve segments which, at serial recordings, recovered within 6 weeks without developing temporal dispersion (increased duration of negative peak of proximal CMAP >30% compared with dCMAP) or conduction slowing (AMAN with reversible conduction failure); and (4) normal or reduced sensory nerve action potential (SNAP) amplitudes not fulfilling the criteria for a diagnosis of AMSAN.5 16 22 24 25 Patients were classified as AMSAN on the basis of the following criteria: (1) acute onset progressive motor weakness of more than one limb and loss of at least one sensory modality; (2) results of motor nerve conduction studies as in AMAN; and (3) SNAP amplitude <50% of LLN in at least two nerves.5 22
The final electrodiagnosis was based on the results of sequential studies and considering the whole electrophysiological history in each patient.
Electrophysiological studies
Motor conduction studies of median, ulnar, peroneal and tibial nerves were performed as previously reported.25 Amplitude and duration of negative peak of dCMAP and CMAP from proximal stimulation, conduction velocities, DMLs and minimal F wave latencies were measured. For sensory conductions, median and ulnar nerves were stimulated at the proximal wrist crease and SNAPs were antidromically recorded by ring electrodes placed at the interphalangeal joints of the third and fifth digit, respectively. In selected patients, median nerves were also stimulated at the elbow, ulnar nerves were stimulated below the elbow and above the elbow and SNAP amplitude ratios were calculated. The sural nerve was stimulated at the lateral malleolus and SNAP recorded orthodromically by a bar electrode placed slightly lateral to the midline in the lower third of the posterior aspect of the leg. SNAPs were obtained by averaging at least eight responses to 1 Hz supramaximal stimuli, latencies were measured to the first deflection from baseline and amplitudes were measured baseline to negative peak. Limb temperature was maintained at 32–34°C. For DML, conduction velocities and F wave latency, we defined the upper and lower limits of normal (ULN and LLN) as mean±2.5 SD of control values of our laboratory. For CMAP and SNAP amplitudes, the LLN was calculated as the mean−2.5 SD of the logarithmically transformed amplitudes of the controls. Normal values for sensory nerve studies are: for median nerve, SNAP amplitude ≥11 μV and sensory conduction velocity (SCV) ≥49 m/s; for ulnar nerve, SNAP amplitude ≥ 10 μV and SCV ≥ 49 m/s; for sural nerve, SNAP amplitude ≥ 6 μV and SCV ≥ 40 m/s. Control values for SNAP amplitude ratios, obtained from 20 median and 20 ulnar nerves of 10 healthy subjects (mean age 34.5 years, range 23–52) are: median, elbow to wrist 0.49±0.07 (range 0.35–0.6); ulnar, below elbow to wrist 0.46±0.09 (range 0.31–0.58); ulnar, above elbow to below elbow 0.78±0.09 (range 0.6–0.93).
Definition of significant changes in serial sensory conduction studies
To evaluate the extent of variation in serial recordings of SNAP amplitudes, we made a test–retest study in 20 volunteers (median age 55 years, range 23–87). In each subject, median, ulnar and sural nerve conductions were repeated 1 week apart by four examiners for a total of 480 recordings. We determined the root mean square per cent coefficient of variation (RMS-%CV) of SNAP amplitude and calculated the least significant change—that is, the minimum difference between two results that can be considered to reflect a true change at a 95% statistical confidence level. The least significant change was calculated using the formula: RMS-%CV×2.77.26 SNAP amplitudes were considered significantly increased or decreased compared with a precedent study when the value varied by at least 44% for the median nerve, 47% for the ulnar nerve and 58% for the sural nerve.
Antiganglioside antibody testing
IgG and IgM to gangliosides GM1, GD1a and GD1b were tested by ELISA as previously described.27 Serum was considered positive when showing antiganglioside antibodies of IgG class with a titre ≥1:400.
Statistical analysis
Two sided Fisher's exact test was employed to explore the association of anti-GD1b antibodies with sensory symptoms, pain, sensory signs and involvement of sensory fibres electrophysiologically demonstrated at baseline and throughout the study. Bonferroni's adjustment for multiple tests was applied and, to obtain an overall α level of 0.05, values of p<0.01 in single tests were considered significant.
Results
Clinical and laboratory findings
From a series of 116 consecutive GBS patients, 13 diagnosed with AMAN and three diagnosed with AMSAN had at least two examinations of motor and sensory conductions in the same nerves within 5 months from onset and an optimal quality of sensory recordings, and were considered for the present study (table 1). Twelve patients were men (75%). Median age was 63 years (range 17–81). All patients presented with symmetric limb weakness.
Clinical features, electrophysiological patterns at final diagnosis and laboratory findings
In the AMAN group, paraesthesia was present in two of 13 (15%) and pain in four of 13 (31%) patients. Deep tendon reflexes (DTR) were absent or hypoactive and absent distally in eight patients, preserved (diffusely hypoactive or normal) in four and brisk in one. One patient had dysphagia; none had respiratory failure or dysautonomia. CSF examination showed albumino-cytological dissociation in 11 patients. Nine patients were treated with intravenous immunoglobulins (IVIg 2 g/kg in 5 days) and the remainders received a course of 3–5 plasmaphereses.
All three AMSAN patients had absent or reduced DTR, paraesthesia in the extremities and glove–stocking sensory loss for all modalities. Patient Nos 14 and 15 had albumino-cytological dissociation at CSF examination and were treated with IVIg. Patient No 14 developed facial diplegia, dysphagia and respiratory failure and died during the third week of illness. Patient No 16 had pain, severe gait ataxia and involvement of the facial and lower cranial nerves requiring a nasogastric tube for feeding. CSF examination 5 days after onset was normal. This patient received a course of five plasmaphereses.
Electrophysiological findings
Baseline electrophysiological examination was performed within 15 days from onset of symptoms (median 7 days; range: 2–15) in at least three motor and three sensory nerves. The second test was performed 2–18 days after the first examination in 13 patients, and 37–146 days after the first examination in three patients. Eight patients received further evaluations. Patient Nos 2, 8, 15 and 16 who were admitted when this study was already in progress, were prospectively studied for motor and sensory conductions. The total number of electrophysiological examinations was 48. The total number of sensory nerves re-examined in subsequent recordings was 47.
Below are the results of the serial electrophysiological studies. For clarity, the electrophysiological findings are presented by group: AMAN with axonal degeneration, AMAN with reversible conduction failure, AMAN with axonal degeneration and minor sensory abnormalities, and AMSAN. Serial sensory conduction studies are reported in full in table 2.
Serial sensory conduction studies in patients with acute motor axonal neuropathy (patient Nos s 1–13) and acute motor and sensory axonal neuropathy (patient Nos 14–16)
AMAN with axonal degeneration
In patient Nos 1–8, baseline electrophysiological examination showed dCMAP amplitudes <80% of LLN in at least two nerves and no signs of demyelination. In subsequent recordings, dCMAP remained stable or further decreased and moderate to abundant fibrillation potentials were found. In patients with long term follow-up, improvement of dCMAP amplitude was seen by weeks 11–32 (median 20 weeks). These patients had normal sensory studies at baseline and were classified as AMAN with axonal degeneration pattern.
In patient Nos 1 and 2, sensory conductions did not substantially change at follow-up. Six patients (patient Nos 3–8) showed significant changes in SNAP amplitude in retested nerves (table 2). In particular, in patient Nos 3 and 4, median nerve SNAP decreased by 50–69% and become abnormal within the 3 weeks after onset (figure 1A). In patient No 5, SNAP amplitude of the right ulnar, median and sural nerves significantly increased within 4 weeks from onset up to 518% of the baseline value (figure 1B). In patients Nos 6 and 7, the amplitude of sural SNAP increased by 207% at day 52 after onset and by 138% at day 102. In patient No 8, left ulnar SNAP amplitude decreased by 64%, 9 days after onset, to recover in 80 days and be increased by 57% compared with baseline on day 191. SNAP amplitude of the left median nerve gradually increased by 59% compared with baseline, and SNAP amplitude of the right median and ulnar nerves was significantly increased by 63–105% on day 191 (figure 1C).

Electrophysiological findings in three patients with acute motor axonal neuropathy and axonal degeneration pattern and normal sensory studies at baseline: (A) patient No 4; (B) patient No 5; and (C) patient No 8. Amplitudes of sensory nerve action potentials (SNAP) and of distal compound muscle action potentials (CMAP) are shown for days after onset. The numbers beside each symbol indicated decreased (−) or increased (+) percentages of SNAP amplitude compared with the baseline evaluation.
AMAN with reversible conduction failure
Patient Nos 9, 10 and 11 showed, at the baseline electrophysiological examination, low amplitude dCMAPs in at least two nerves, mildly prolonged DML in six nerves (median 122% of ULN; range 109–132) and partial motor CB in nine nerves. MCV was normal except for the site of CB in five nerves. Within 6 weeks, amplitude and latency of dCMAPs normalised, and CB and conduction slowing at the site of CB resolved without development of excessive temporal dispersion of CMAPs. At follow-up, little or no signs of denervation were seen at EMG. These three patients, classified as AMAN with reversible conduction failure pattern, had normal sensory studies at the first examination performed within the first week.16 23
In the left median nerve of patient No 9, SNAP amplitude increased by 220% in 8 weeks along with normalisation of dCMAP amplitude, whereas proximal SNAP amplitude increased by 139% along with improvement of proximal CMAP amplitude and disappearance of motor CB (figure 2). At day 23, because of the greater increase in SNAP amplitude from wrist stimulation compared with SNAP from elbow stimulation, the SNAP ratio decreased from 0.75 to 0.42. The SNAP ratio increased to 0.56 at day 59. In the remaining two patients, no significant changes were observed in SNAPs, and SNAP ratios remained within the normal range throughout the follow-up.

Patient No 9, left median nerve. Upper tracings: serial recordings of compound muscle action potential from the abductor pollicis brevis muscle after stimulation at the wrist and elbow. Lower tracings: serial recordings of sensory nerve action potential from digit III after wrist and elbow stimulation.
AMAN with axonal degeneration and minor sensory abnormalities
Patient Nos 12 and 13 had an axonal degeneration pattern in motor fibres, no sensory signs and minor baseline abnormalities of sensory conductions not fulfilling the criteria for AMSAN.
In patient No 12, baseline electrophysiological examination showed normal sensory conduction in the right ulnar nerve, abnormal right median SNAP amplitude (70% of LLN) and abnormal SNAP amplitude and SCV in the right sural nerve (88% and 90% of LLN, respectively). Nine days later, SNAP amplitude increased by 51% becoming normal in the sural nerve and was significantly increased (by 239%) in the ulnar nerve.
In patient No 13, who had only abnormal median SNAP amplitude and SCV at baseline (73% and 88% of LLN, respectively), re-examination 5 months after onset showed, in the median nerve, the persistence of low amplitude SNAP but normalisation of SCV whereas there was a significant decrease in SNAP amplitude in the sural nerve (−61%) and a non-significant decrease in SNAP amplitude in the ulnar nerve (by 42%). At 9 months, median nerve SNAP was increased by 128% compared with baseline becoming normal.
AMSAN
Three patients (patient Nos 14–16) had dCMAP amplitudes <80% of LLN in at least two nerves, SNAP amplitude <50% of LLN in at least two nerves and were classified as AMSAN.
Patient No 14 showed no significant changes in motor and sensory conductions during the first 2 weeks.
In No patient 15, baseline CMAP amplitudes were markedly decreased in both median and ulnar nerves and at the LLN in the peroneal nerves whereas SNAP amplitudes were <50% of LLN in both ulnar nerves and normal in both median nerves and in the right sural nerve. At follow-up, distal CMAP disappeared or further decreased in amplitude in the upper limb nerves, remained stable in the left peroneal nerve and improved by 59% in the right peroneal nerve by day 55. Conversely, SNAP amplitude of the ulnar nerves significantly improved by 150–300% and normalised within 1 week during IVIg administration (figure 3).

Patient No 15, left ulnar nerve. Upper tracings: serial recordings of compound muscle action potential from abductor digiti minimi muscle after stimulation at the wrist, below the elbow and above the elbow. Lower tracings: serial recordings of sensory nerve action potential recorded from digit V after wrist stimulation.
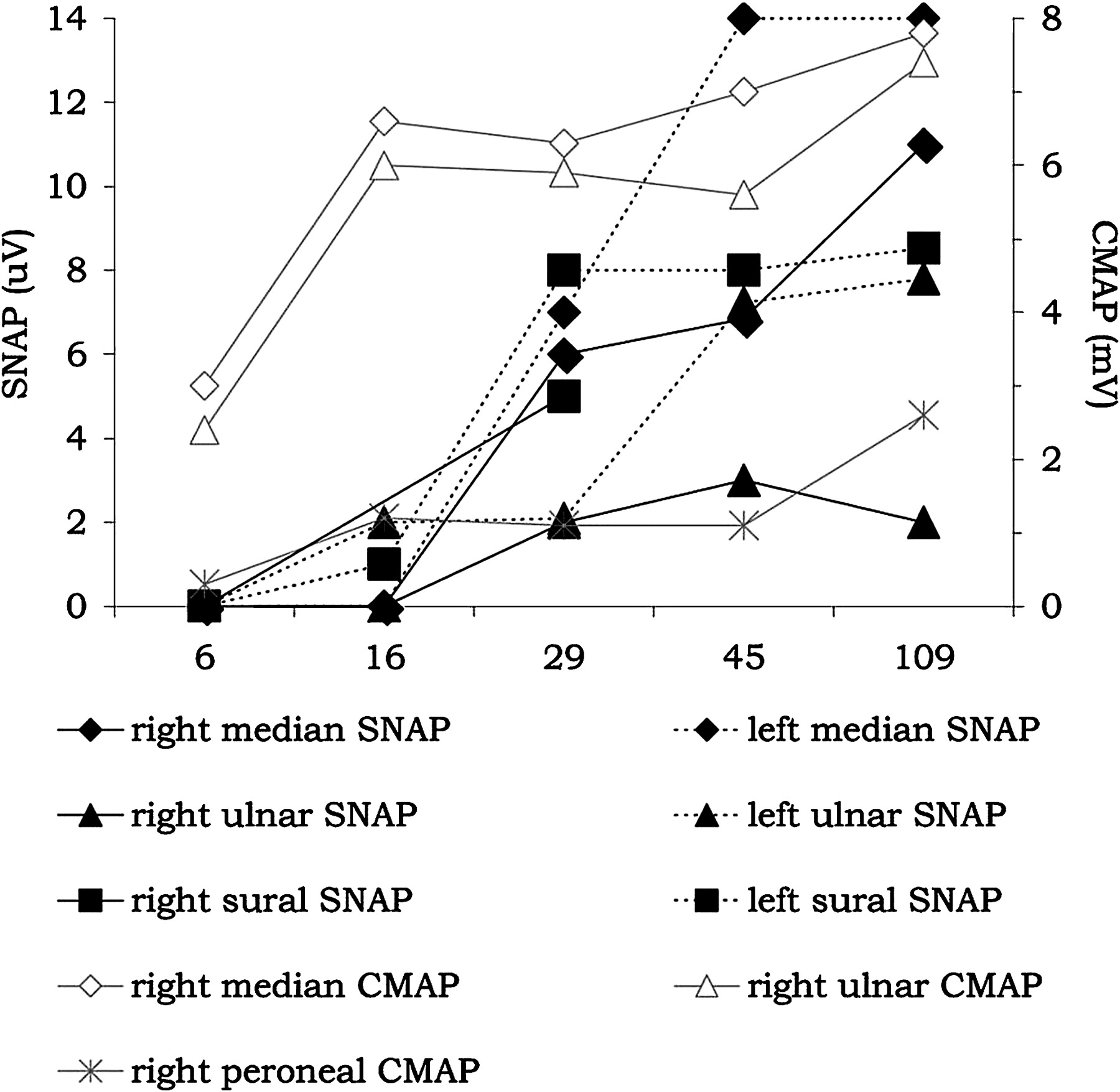
In patient No 16, baseline electrophysiological examination showed low amplitude distal CMAP in the right median, ulnar and peroneal nerves, and a partial motor CB in the forearm segment of the right ulnar nerve. DMLs and MCVs were normal. SNAPs were not recordable in the median, ulnar and sural nerves bilaterally. Within 4 weeks, distal CMAP amplitudes normalised in the upper limb nerves and partial CB resolved in the right ulnar nerve without development of excessive temporal dispersion of proximal CMAP, indicating reversible conduction failure in motor fibres. During follow-up, SNAPs reappeared early in all nerves and finally normalised in three nerves (figure 4).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Serial electrophysiological findings in patient No 16 with acute motor and sensory axonal neuropathy and reversible conduction failure pattern. Amplitudes of distal compound muscle action potentials (CMAP) and of sensory nerve action potentials (SNAP) are shown for days after onset.
Summary of serial sensory conduction findings
In the AMAN group, one sensory nerve showing minor abnormalities at baseline significantly recovered at follow-up, and 11 of 32 initially normal sensory nerves (34%) of six patients (five with baseline normal sensory conductions, one with minor sensory abnormalities at baseline) showed an increase in SNAP amplitude (median increase by 106%, range 57–518%). Conversely, in three initially normal nerves of three AMAN patients (two with baseline normal sensory conductions, one with minor sensory abnormalities at baseline) a decrease in SNAP amplitude was found (median decrease by 60%, range 50–69%). In summary, in the AMAN group, we found that: (1) two patients had minor sensory changes in three nerves at baseline; (2) 14 of 32 initially normal nerves (44%) and seven of 11 patients with baseline normal sensory conductions (64%) showed significant changes in SNAP amplitude; and (3) overall sensory fibres were involved in 49% of nerves and in 69% of AMAN patients.
In one AMSAN patient, SNAP amplitude increased by 150–300% in two nerves. In six nerves of another AMSAN patient, SNAPs were unrecordable at baseline but reappeared during follow-up, normalising in three nerves.
Overall, 11 of 16 patients with axonal GBS (69%) showed significant changes in SNAP amplitudes in 23 of 47 (49%) retested nerves (tables 1 and 2).
Antiganglioside antibodies
Antibodies to at least one of the examined gangliosides were found in 13 of 15 tested patients (87%) (table 1). There was no difference in the immunological profile of AMAN and AMSAN patients. Anti-GD1b antibodies were not associated with paraesthesia (p=0.026), pain (p=0.315), sensory loss (p=0.077), electrophysiological involvement of sensory fibres at baseline (p=0.119) or at follow-up (p=1.000).
Discussion
Axonal GBS is a term derived from pathology but electrophysiologically defined axonal GBS subtypes are not only the expression of axonal degeneration. Three AMAN and one AMSAN patient in this study showed reversible conduction failure in motor fibres at serial recordings. Reversible conduction failure, described in some patients with antiganglioside antibodies and in a GBS subtype named acute motor conduction block neuropathy, is ascribed to an antibody mediated reversible conduction impairment at the axolemma of the nodes of Ranvier.16 28 According to experimental studies, antiganglioside antibodies attack primarily the axolemma of motor fibres at the nodes of Ranvier, sequentially inducing complement activation, disruption of sodium channel clusters and nodal architecture, and finally Wallerian-like degeneration.15 In some patients, this pathological cascade may arrest to the stage of sodium channel derangement, resulting in transient and reversible failure of axonal function.2 The occurrence of reversible conduction failure in motor fibres has not been reported in AMSAN before and may explain why some AMSAN patients do not have a poor outcome.
The 13 patients with AMAN that we reported had clinically pure motor GBS with no sensory signs. DTR were preserved in four patients and brisk in one. According to Asbury and Cornblath, universal areflexia is the rule in GBS although distal areflexia with definite hyporeflexia of the biceps and knee jerks will suffice for diagnosis if other features are consistent.29 DTR may be preserved throughout the disease course in patients with AMAN.30 Moreover, 48% of Chinese and 33% of Japanese patients with AMAN showed hyperreflexia in the recovery phase.3 16 Moderate weakness and the normal afferent branch of the myotatic reflex arch could account for the preservation of DTR in our patients but they do not explain hyperreflexia. Electrophysiological evidence of lower motor neuron hyperexcitability has been reported in some patients with AMAN with hyperreflexia.31 32 In conclusion, preserved DTR and even hyperreflexia may occur in patients with AMAN and are not inconsistent with the diagnosis. Paraesthesia was present in 15% and pain in 31% of patients. Two patients (15%) had mild sensory abnormalities at the baseline electrophysiological examination. As for preserved DTR, these features are not inconsistent with AMAN diagnosis. Paraesthesia has been reported in 10–40% of patients and pain, even severe, accompanies AMAN in up to 66% of patients.3 10 11 33 The sensory conduction abnormalities we found at baseline in two patients did not satisfy the electrophysiological criteria for defining AMSAN and have been reported in 5–12% of AMAN patients.3–5 11 22
Serial recordings of sensory conductions demonstrated that sensory fibres were subclinically involved in most AMAN patients, even when baseline sensory conductions appeared normal. In fact, SNAP amplitudes significantly increased in 34% of initially normal nerves of six patients indicating that, at baseline, sensory fibres were involved although not enough to produce a decrease in SNAP below the LLN. Similarly, in eight nerves of two AMSAN patients, initially absent or reduced SNAPs recovered at follow-up. In three AMAN patients, SNAP amplitude decreased in three initially normal nerves, indicating later involvement of sensory fibres. Overall, in our series, baseline and subsequent serial recordings allowed the detection of sensory fibre involvement in 49% of nerves and in 69% of AMAN patients.
The improvement in SNAPs followed two courses. In five nerves of three AMAN and in eight nerves of two AMSAN patients, SNAP amplitudes increased rapidly (within 2–4 weeks from onset). Similar features have been reported in one AMAN patient with reversible conduction failure pattern and antibody to GM1/GalNAc-GD1a complex and in patients with acute sensory ataxic neuropathy and antibodies to GD1b, suggesting that reversible conduction failure may occur not only in motor but also in sensory fibres.34 35 In other patients, SNAP amplitudes increased over months according to an axonal regeneration process.
We did not find different immunological profiles in electrophysiologically pure motor GBS, AMAN with minor sensory involvement or AMSAN. In particular, we could not confirm the hypothesis that anti-GD1b antibodies were associated with clinical or electrophysiological involvement of sensory fibres.
Although our findings demonstrate that AMAN is not an exclusively motor neuropathy, there is no doubt that the involvement of motor fibres is predominant and determinant for clinical severity. The bases of this phenomenon are not clear. Biochemical studies demonstrated that sensory and motor nerves express similar quantities of GM1 and GD1a gangliosides.36 37 However, serum from an AMAN patient with anti-GD1a antibodies selectively stained nodes of Ranvier of motor fibres and an anti-GD1a monoclonal antibody preferentially stained motor spinal roots in humans and rodents.37 38 A study employing several monoclonal antibodies and GD1a derivates suggested that both fine specificity of antibodies and different orientation/exposure of ganglioside in motor and sensory fibres contribute to target recognition by antiganglioside antibodies.39 Moreover, a number of biophysical differences between human sensory and motor axons have been reported. Sensory axons have less supernormality and late subnormality, longer strength–duration time constant and a lower rheobase than motor nerves.40–43 Sensory axons accommodate more than motor axons to longlasting hyperpolarising currents, suggesting greater expression of the hyperpolarisation activated inward rectifier channels and greater expression of persistent Na+ channels.41 44 Lastly, the electrogenic Na+–K+ ATPase is more active in sensory nerves.45 These differential biophysical features may explain the fact that motor fibres are more affected than sensory fibres by the attack of the same antibody and the coexistence of positive sensory symptoms such as paraesthesias in some AMAN patients.
Whatever the reason for the differential involvement of motor and sensory fibres in axonal GBS subtypes, this study shows that: (1) sensory fibres are involved in AMAN although to a lesser extent than motor fibres and not enough to produce a clinical correlate; (2) reversible conduction failure is present in motor and sensory fibres both in AMAN and AMSAN; and (3) AMAN and AMSAN, having in common a immunological profile, electrophysiological features and pathophysiology, are a continuum, with AMSAN with axonal degeneration representing the more severe end of the spectrum.