Article Text

Abstract
Background Frontotemporal dementia-amyotrophic lateral sclerosis (FTD-ALS) is a heritable form of FTD, but the gene(s) responsible for the majority of autosomal dominant FTD-ALS cases have yet to be found. Previous studies have identified a region on chromosome 9p that is associated with FTD and ALS.
Methods The authors report the clinical, volumetric MRI, neuropathological and genetic features of a new chromosome 9p-linked FTD-ALS family, VSM-20.
Results Ten members of family VSM-20 displayed heterogeneous clinical phenotypes of isolated behavioural-variant FTD (bvFTD), ALS or a combination of the two. Parkinsonism was common, with one individual presenting with a corticobasal syndrome. Analysis of structural MRI scans from five affected family members revealed grey- and white-matter loss that was most prominent in the frontal lobes, with mild parietal and occipital lobe atrophy, but less temporal lobe atrophy than in 10 severity-matched sporadic bvFTD cases. Autopsy in three family members showed a consistent and unique subtype of FTLD-TDP pathology. Genome-wide linkage analysis conclusively linked family VSM-20 to a 28.3 cM region between D9S1808 and D9S251 on chromosome 9p, reducing the published minimal linked region to a 3.7 Mb interval. Genomic sequencing and expression analysis failed to identify mutations in the 10 known and predicted genes within this candidate region, suggesting that next-generation sequencing may be needed to determine the mutational mechanism associated with chromosome 9p-linked FTD-ALS.
Conclusions Family VSM-20 significantly reduces the region linked to FTD-ALS on chromosome 9p. A distinct pattern of brain atrophy and neuropathological findings may help to identify other families with FTD-ALS caused by this genetic abnormality.
- Frontotemporal dementia
- amyotrophic lateral sclerosis
- chromosome 9p
- TAR-DNA binding protein 43
- ALS
- clinical neurology
- dementia
- genetics
- pathology
Statistics from Altmetric.com
- Frontotemporal dementia
- amyotrophic lateral sclerosis
- chromosome 9p
- TAR-DNA binding protein 43
- ALS
- clinical neurology
- dementia
- genetics
- pathology
Introduction
Clinically and pathologically, there is considerable overlap between frontotemporal dementia (FTD) and the amyotrophic lateral sclerosis (ALS) type of motor neuron disease,1 and many FTD patients display concurrent ALS symptoms, leading to a combined diagnosis of FTD with ALS (FTD-ALS). Individuals with FTD-ALS usually display more aggressive disease than other clinical FTD syndromes, with a median survival of approximately 1.5–3 years after diagnosis.2 3 On structural MRI scans, FTD-ALS is associated with frontal and temporal lobe atrophy that is more severe than that found in cognitively normal ALS patients4 but less extensive than related forms of FTD without ALS.5 At autopsy, most FTD-ALS cases are found to have neuronal cytoplasmic inclusions that are immunoreactive for both ubiquitin and the transactive response (TAR) DNA-binding protein with Mr 43kDa (TDP-43), classified as FTLD-TDP type 3 (Mackenzie classification6 7 or type 2 according to Sampathu et al).8
FTD-ALS is a heritable form of FTD,9 but in the majority of FTD-ALS cases the causative gene has yet to be identified. A locus on chromosome 9 in the 9p21.2–p13.3 region, first described in two independent reports by Vance et al. and Morita et al. in 2006,10 11 is likely to contain a gene responsible for a substantial proportion of autosomal dominant FTD-ALS cases. A number of large, independent FTD-ALS kindreds have since been linked to a similar chromosome 9p region,12–14 defining a shared candidate region of 7.7 Mb containing 52 candidate genes.15
Here we describe a new FTD-ALS family definitively linked to chromosome 9p with novel clinical, neuroimaging and neuropathological findings whose disease-associated haplotype reduces the size of the candidate region linked to FTD-ALS to a 3.7 Mb interval on chromosome 9p containing only 10 genes.
Methods
Subjects
All FTD and/or ALS family members from whom genetic samples were obtained were evaluated at the Mayo Clinic (Rochester, Minnesota), University of British Columbia (UBC) and/or the University of California, San Francisco (UCSF). Since family members were evaluated at all three locations, the family was named VSM-20 for ‘Vancouver, San Francisco and Mayo family 20.’ VSM-20 individuals were designated as affected if they met Neary et al16 criteria for behavioural variant FTD (bvFTD) or El Escorial Criteria for ALS,17 and had a clinical dementia rating (CDR)18 sum of boxes (SB) score of greater than 1.0. This study was approved by the Institutional Review Boards and ethics committees at the Mayo Clinic, UBC and UCSF. Written informed consent was obtained from all patients (or designated surrogates) participating in the study.
MRI scans
Imaging data from VSM-20 members were compared against two groups of controls. One group was composed of sporadic bvFTD cases with high-quality MRI scans performed at either Mayo Clinic or UCSF within 3 months of clinical evaluation. These were matched to VSM-20 family members based on CDR-SB scores as well as on which scanner data were acquired. A second (normal) control group was composed of subjects with normal neurological and neuropsychological examinations, and CDR-SB scores of 0.
MRI scans were obtained on a 1.5 T Magnetom VISION system or 3 T Trio Tim (Siemens, Iselin, New Jersey) systems at UCSF and a 3 T GE Signa HDxt system at the Mayo Clinic using high-resolution T1-weighted 3D magnetisation prepared rapid acquisition gradient echo (MPRAGE) sequences. For the 3 T scans at UCSF, the scan parameters were: TR/TE/T1, 2300/3/900 ms, flip angle 9°, 26 cm field of view (FOV), 256×256 in plane matrix, with a phase FOV of 0.94 and slice thickness of 1.0 mm. Scan parameters for the other two scanners are described in previous publications.19 20
Volumetric MRI analysis
Cortical reconstruction and volumetric segmentation were performed with the Freesurfer image analysis suite (documented and freely available at http://surfer.nmr.mgh.harvard.edu/). The technical details of these procedures are described in a prior publication.21 A univariate ANOVA with Tukey post-hoc test, controlling for the scanner on which scans were acquired and age at MRI scan, was used to compare the mean lobar volumes and region of interest grey-matter volumes from each group. Statistical analyses were performed in SPSS for Windows (version 17; SPSS, Chicago, Illinois).
Linkage analyses and delineation of candidate region
We carried out a 4 cM genome-wide scan using DNA samples of 19 individuals from family VSM-20 (six patients, two spouses and 11 at-risk individuals; figure 1). Short tandem repeat (STR) genotyping for 1091 markers was done by deCODE genetics. We calculated linkage analysis with the assumption of an autosomal dominant mode of inheritance and a disease frequency of 0.01%. A cumulative risk curve was calculated using mean age at onset in the family and assumption of maximal disease penetrance of 90% at age ≥60 years, and nine penetrance classes were defined. All patients with FTD, ALS or FTD-ALS were considered affected in the analysis. Two-point Logarithm of the odds (LOG) scores were calculated using MLINK from the FASTLINK package assuming equal recombination rates for males and females.
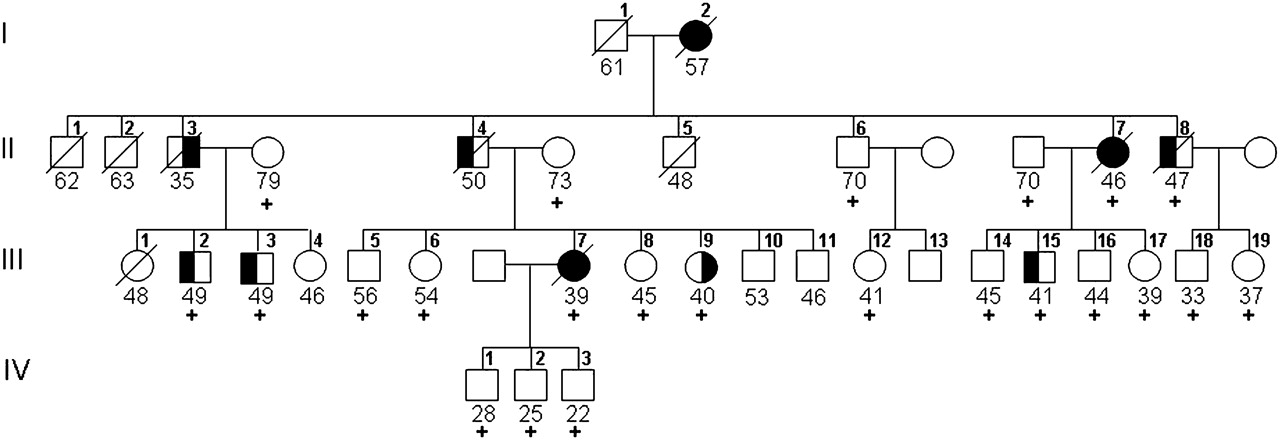
Pedigree of family VSM-20. Black symbols represent patients affected with behavioural-variant frontotemporal dementia (left side filled), amyotrophic lateral sclerosis (right side filled) or both. White symbols represent unaffected individuals or at-risk individuals with unknown phenotype. The Roman numeral to the left of the pedigree denotes the generation; Arabic numbers above the individuals denote individuals. The Arabic numbers below the individuals denote age at onset for patients and either age at last examination or age at death for unaffected or at-risk individuals with unknown phenotype. The ‘+’ sign indicates individuals included in the linkage analysis. For generation IV, only individuals included in the linkage analysis are shown.
For genetic fine mapping, we selected an additional set of 10 STR markers spanning a 33.1 cM region from the Marshfield sex-average linkage map and the genetic map of the Cooperative Human Linkage Center for genotyping in all members of family VSM-20 (seven patients, three spouses and 13 at-risk individuals). DNA was PCR amplified using one fluorescently labelled primer and analysed on an automated ABI3730 DNA-analyser (Applied Biosystems, Foster City, California). Allele identification and scoring was performed using GENESCAN and GENOTYPER software (Applied Biosystems) and normalised to the CEPH genotype database, except for D9S251 and D9S304 for which fragment sizes were not available.
Mutation analyses
In family VSM-20, a genomic DNA (gDNA) sequencing analysis was performed for all 10 candidate genes located within the 3.7 Mb minimal candidate region defined by all published 9p-linked families and 13 additional genes from the larger candidate region defined by family VSM-20 alone. Mutation analysis of all exons of GRN and TARDBP was also performed. For the five protein coding genes in the minimal candidate region, we further searched for complete genomic deletions or duplications using quantitative SYBRGreen real-time PCR. Complementary (cDNA) mutation analysis was performed for MOBKL2B and c9orf72. Details on the mutation analyses can be found in supplementary genetics methods and supplementary tables S1 and S2.
Neuropathological analysis
A post-mortem examination was performed on three affected family members (cases III-7, II-8 and II-7). Immunohistochemistry (IHC) was performed using previously published methods22 with primary antibodies that recognise ubiquitin, p62, hyperphosphorylated tau, α-synuclein, Aβ, FUS and TDP-43. Details on the neuropathological analysis can be found in supplementary neuropathology methods.
Results
Clinical syndromes in family VSM-20
The clinical features of 10 family members who met research criteria for bvFTD and/or ALS are summarised in table 1. The family is of Irish ancestry. The mean age of onset was 45.7±7.0 years. The mean duration of symptoms was 5.4±3.0 years. Three individuals had a bvFTD phenotype with no clear evidence of motor impairments, while two family members had bvFTD with mild Parkinsonism (primarily elevated tone) on physical exam. Two individuals had a pure limb-onset ALS phenotype with minimal cognitive or behavioural impairments. Three individuals displayed a combination of bvFTD symptoms and ALS, with one individual presenting with apraxia and Parkinsonism consistent with a corticobasal syndrome (CBS, III-7). Detailed descriptions of examples of each of the three main phenotypes are included as supplementary case descriptions.
Clinical heterogeneity in affected members of family VSM-20
Patterns of brain atrophy in VSM-20 compared with sporadic bvFTD and controls
Lobar brain volumes were derived in five affected family members (four predominantly bvFTD (II-8, III-2, III-3, and III-15), one predominantly ALS (III-9)) as compared with 10 sporadic bvFTD cases, matched for CDR-SB scores to the family members, and 13 normal control subjects. Atrophy was more extensive in VSM-20 members with isolated bvFTD (II-8, figure 2A) as compared with the family member with pure ALS (III-9, figure 2B).

Patterns of brain atrophy in two VSM-20 clinical phenotypes: coronal T1-weighted MRI section at MNI coordinate y=14, and Freesurfer-derived cortical thickness maps in (A) a 54-year-old behavioural-variant frontotemporal dementia subject (II-8) as compared with 20 male normal control subjects (mean age 55.5) and (B) a 40-year-old pure amyotrophic lateral sclerosis subject (III-9) as compared with 25 female normal control subjects (mean age 59.8) displayed on rendered normal control MRI template. Coloured areas indicate cortical thickness reductions p<0.05 to p<0.001, uncorrected, versus controls.
Cortical volumes were reduced in the VSM-20 family members and the sporadic bvFTD group as compared with controls (figure 2, table 2). This volume loss was reflected in decreased cortical grey- and white-matter volume in VSM-20, but only decreased grey-matter volume in the sporadic bvFTD group. The patterns of cortical volume loss were also different in the VSM-20 members as compared with the sporadic bvFTD group. Whereas both groups displayed bilateral frontal lobe atrophy as compared with controls, only the sporadic bvFTD displayed temporal lobe atrophy. In contrast, only the VSM-20 family members displayed subtle parietal and right occipital lobe atrophy as compared with controls (table 2).
Patterns of brain atrophy in affected VSM-20 family members
Neuropathology
A detailed summary of neuropathological data from the three affected family members (II-7, II-8 and III-7) is provided in supplemental table S3. All three cases showed similar pathology that varied only in severity. The brain weights were reduced (mean weight=1050 g) and showed consistent atrophy of the frontal lobes but variable involvement of temporal and parietal regions (figure 3A).

Post-mortem neuropathology in affected members of VMS-20. Cerebral atrophy was most consistent in the frontal lobes (A, case II-8). The corticospinal tracts showed loss of myelin staining (B, case III-7). Ubiquitin- and TDP-43-immunoreactive neuronal cytoplasmic inclusions (NCI) were present in all layers of the frontal and temporal neocortex (arrows, C) and either had a smooth round contour or appeared to be an aggregate of coarse granules (D). TDP-43-positive granular preinclusions were most common in the upper neocortical layers (E). Swollen dystrophic neurites in the neocortex were best demonstrated with ubiquitin immunoistochemistry (F). Moderate numbers of TDP-43-immunoreactive glial cytoplasmic inclusions were present in the subcortical white matter (G). In the hippocampus, compact neuronal cytoplasmic inclusions (NCI) were numerous in the dentate granule cells (H), while hippocampal pyramidal neurons often contained more ill-defined NCI (I). NCI were present in lower motor neurons of the brainstem and spinal cord, and had a granular, filamentous (J) or compact (K) morphology. There were numerous round NCI and neurites in the cerebellar granular layer that were immunoreactive ubiquitin but not TDP-43 (L). (A) gross photograph; (B) haematoxylin and eosin with luxol fast blue; (C, F, H–L), ubiquitin immunohistochemistry; (D, E and G) TDP-43 immunohistochemistry. Scale bar: (A) 6 cm; (B) 300 μm; (C, F, H and I) 50 μm; (D, E and G) 12 μm; (J and K) 30 μm; (L) 20 μm.
Affected regions of cerebral cortex showed chronic degenerative changes with neuronal loss, reactive gliosis and superficial laminar spongiosis. The corticospinal tracts showed decreased myelin staining at all levels of brainstem and spinal cord, most notably in case III-7 (figure 3B). IHC for ubiquitin, p62 and TDP-43 demonstrated various types of neuronal cytoplasmic inclusions (NCI) in all neocortical layers (figure 3C). The predominant morphology was dense round bodies, approximately the size of the nucleus, with either a smooth contour or an appearance of aggregated coarse granules (figure 3D). In addition, layer II neocortex contained many neurons with diffuse cytoplasmic granular reactivity for TDP-43 (preinclusions) (figure 3E). Swollen dystrophic neurites were moderately common in deeper cortical layers and were better demonstrated with ubiquitin and p62 IHC than with TDP-43 (figure 3F). Rare TDP-43-immunoreactive glial cytoplasmic inclusions (GCI) were present in the subcortical white matter (figure 3G). These pathological changes were present in all neocortical regions examined, with the frontal and temporal lobes more affected than parietal and occipital.
Both compact and granular ubiquitin/p62/TDP-43-positive NCI were numerous in dentate granule cells of all cases (figure 3H). Hippocampal pyramidal neurons often contained ill-defined aggregates of fine granules that were ubiquitin- and p62-immunoreactive but only rarely TDP-43-positive (figure 3I). Lower motor neurons in the brainstem and spinal cord frequently contained NCI that were reactive for ubiquitin, p62 and TDP-43 (figure 3J, K). Finally, the granule cell layer of the cerebellar cortex contained numerous small round NCI and short neurites that were only demonstrated with ubiquitin and p62 IHC (figure 3L). Neuronal intranuclear inclusions were not identified in any of the cases.
IHC for tau, Aβ, α-synuclein and FUS proteins did not show any specific pathological changes beyond those expected for patient age. Specifically, ubiquitin/p62-immunoreactive, TDP-43-negative NCI and neurites did not label for any of these other proteins.
Genome-wide linkage analysis and identification of minimal linked region
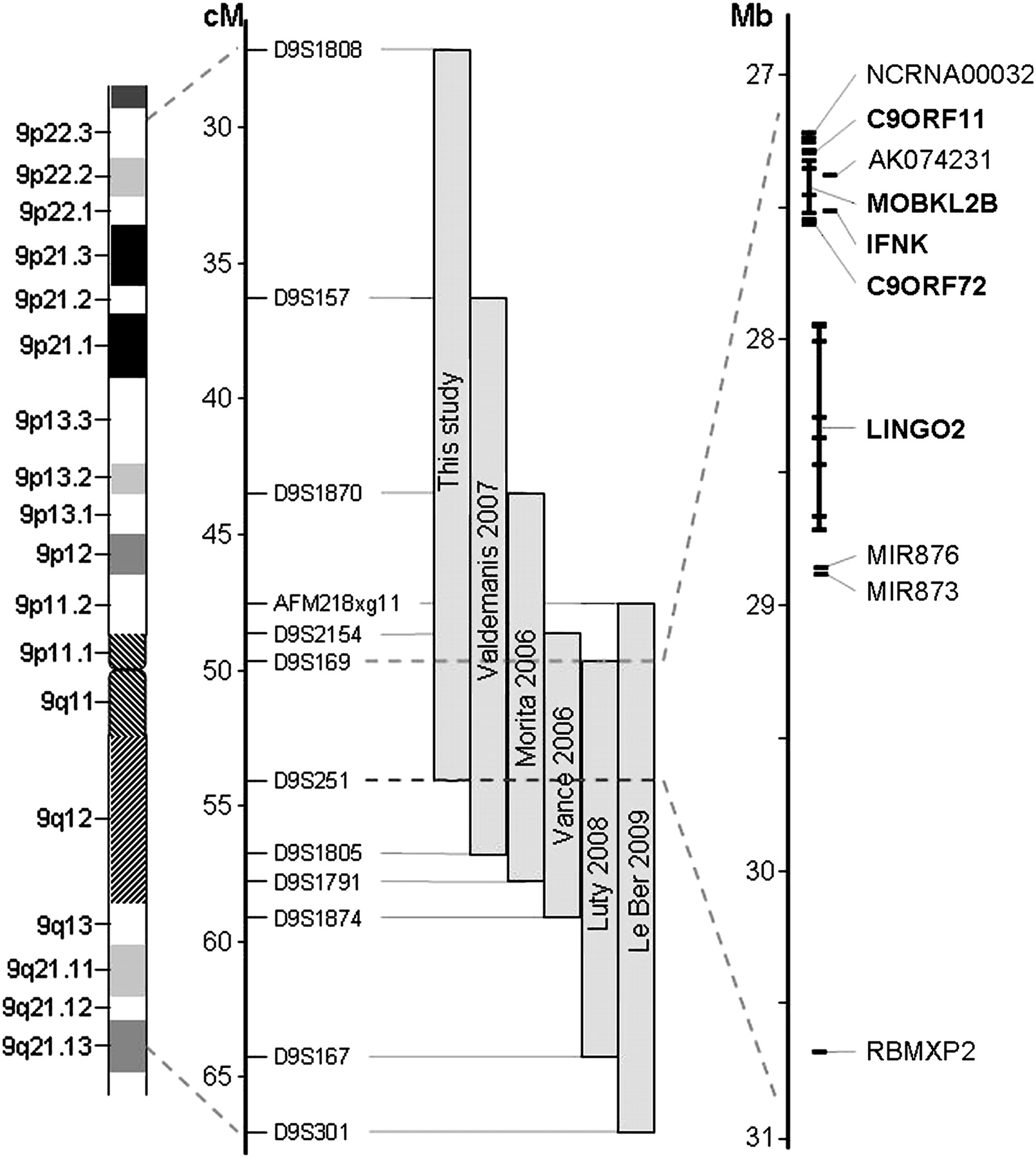
Genome-wide linkage analysis in family VSM-20 identified only one region of suggestive linkage at chromosome 9p, with a maximum two-point LOD-score of 2.59 at D9S1870 (θ=0). A total of six flanking STR markers from the genome scan also showed LOD-scores >1. To fine-map the candidate region at 9p, we analysed six STR markers from the genome scan and an additional 10 STR markers in a 33.1 cM interval flanked by markers D9S1808 and D9S1817 in all family members and reanalysed genotypes. At this time, individual III-15 was re-evaluated and found to meet clinical criteria for bvFTD and therefore was considered affected in all subsequent analysis. The highest two-point LOD-score, 3.01, was obtained with marker D9S1870 in the absence of recombinants (table 3). A disease haplotype was present in all patients, and obligate recombinants defined a candidate region of 28.3 cM corresponding to 14.2 Mb between markers D9S1808 and D9S251 (figure 4). This candidate region overlaps with the previously reported region linked to the combined phenotype of FTD and ALS on chromosome 9p10–15 and significantly reduces the candidate region to a 3.7 Mb interval between D9S169 and D9S251, containing 10 known and predicted genes (figure 5).
Two-point LOD scores for 9p markers in family VSM-20
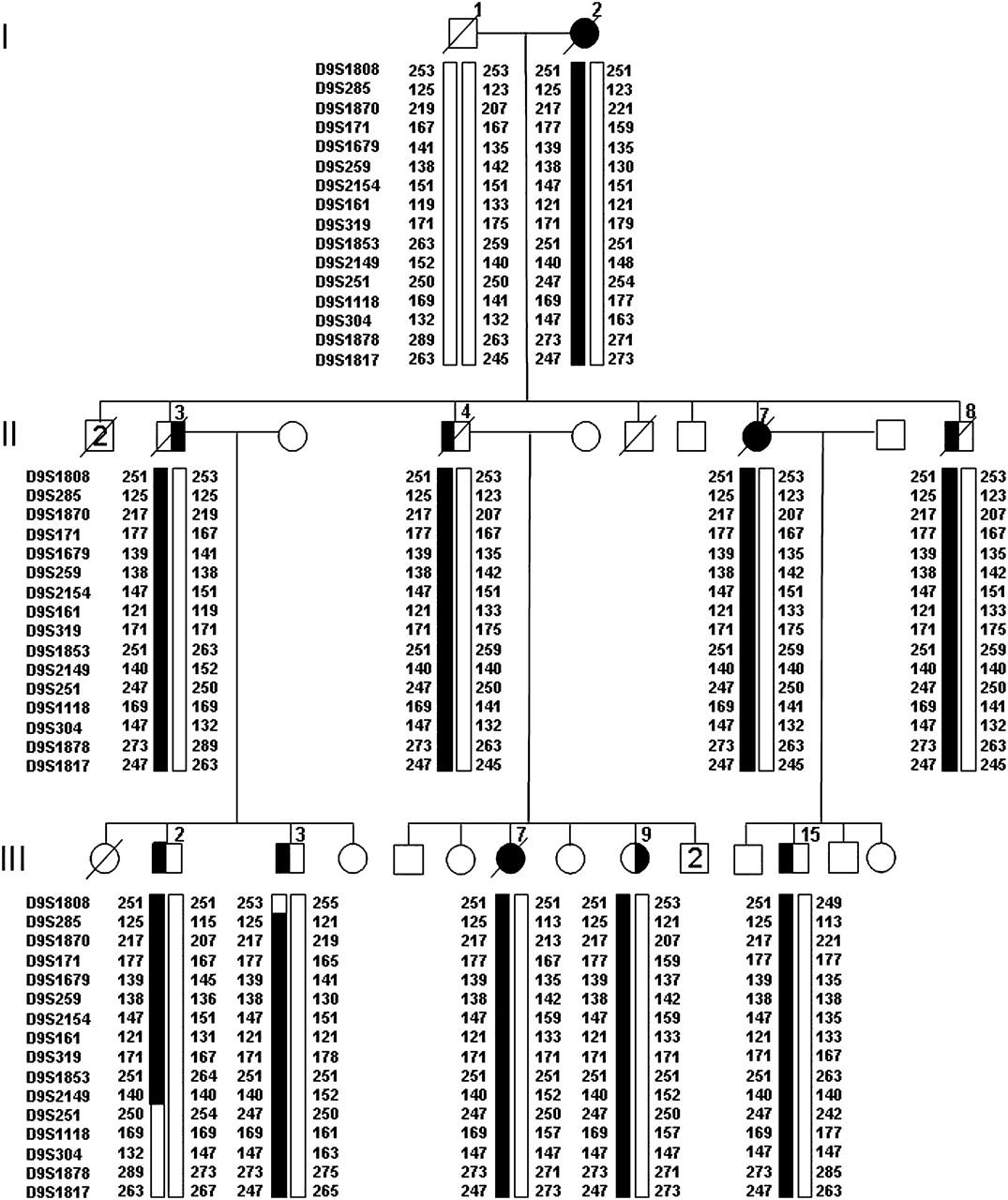
Disguised linkage pedigree of family VSM-20. Black symbols represent patients affected with behavioural-variant frontotemporal dementia (left side filled), amyotrophic lateral sclerosis (right side filled) or both. White symbols represent unaffected individuals or at-risk individuals with unknown phenotype. The Roman numeral to the left of the pedigree denotes the generation; Arabic numbers above the individuals denote individuals. Haplotypes are based on 16 informative markers at chromosome 9p. Haplotypes for individuals I-1, I-2, II-3 and II-4 are inferred from genotype data of siblings and offspring. Detailed pedigree is shown in Figure 1.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
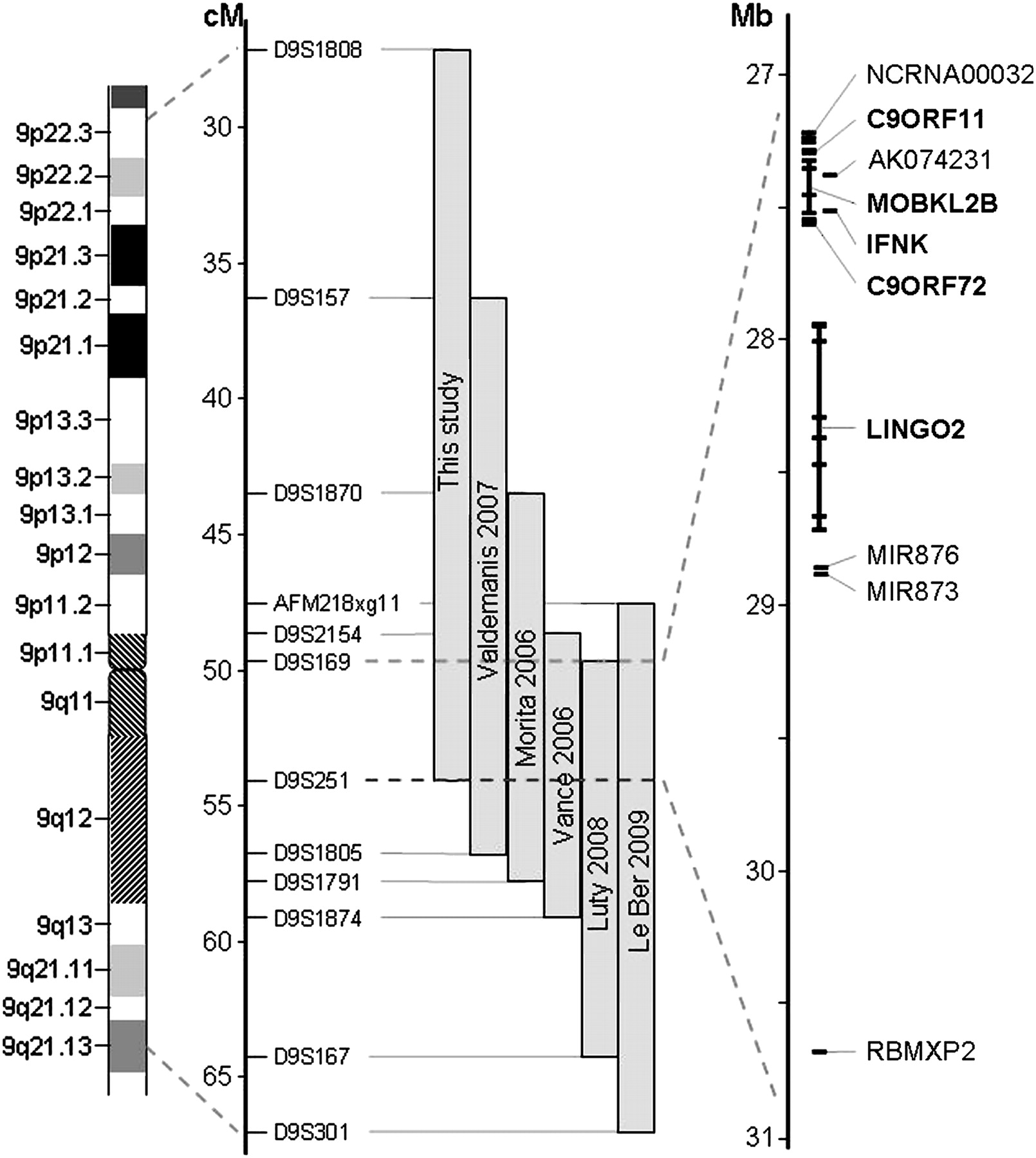
Schematic presentation of chromosome 9p FTD-ALS locus. Grey bars indicate the minimal candidate regions on chromosome 9p in family VSM-20 and all other previously reported 9p-linked FTD-ALS families. Together, these studies define a 3.7 Mb interval between D9S169 and D9S251 containing 10 genes (shown in detail on the right). The five protein coding genes analysed in detail in this study are highlighted in bold.
Mutation analysis of candidate genes
Bioinformatic analysis of the 3.7 Mb minimum candidate interval identified five protein coding genes (c9orf11, MOBKL2B, IFNK, c9orf72 and LINGO2), two micro RNA genes (miR-873 and miR-876), a large non-protein coding RNA gene (NCRNA00032) and two predicted genes (AK074231 and RBMXP2) (figure 5). No mutations were identified by direct gDNA sequencing in the exons (coding and non-coding), flanking intronic sequences and 3′ untranslated regions of the five protein coding genes or in the predicted exons of the other five genes. Furthermore, cDNA splicing analysis of the two genes with multiple coding exons and expression in cerebellum (MOBKL2B and c9orf72) did not identify altered splicing patterns or small deletions or duplications in the RT-PCR products. Mutations in the coding exons of an additional 13 genes located within the larger candidate region identified in family VSM-20 alone were also excluded by gDNA sequencing (supplemental table S4). Pathogenic mutations in GRN and TARDBP were also excluded.
Analysis of three affected members of family VSM-20 on the Affymetrix genome-wide human SNP array 6.0 did not identify differences in gene dosage within the chromosome 9p linked region. In addition, complete genomic deletions or multiplications of c9orf11, MOBKL2B, IFNK, c9orf72 and LINGO2 were excluded by quantitative real-time PCR.
Discussion
In the past 3 years, six groups have reported families with autosomal dominant FTD-ALS with significant or suggestive genetic linkage to chromosome 9p.10–15 Here we report on a novel autosomal dominant FTD-ALS family with conclusive evidence for linkage to chromosome 9p. Similar to other chromosome 9p-linked FTD-ALS families, affected individuals in this family had isolated bvFTD, ALS or a combination of both clinical syndromes.10 11 However, other aspects of the VSM-20 phenotype, including Parkinsonism, were different from previously described 9p-linked families. Moreover, neuroimaging and neuropathological findings implicate a role for white matter and glial abnormalities in the pathophysiology of disease in this family. A consistent and unique aspect of the neuropathology is the presence of some NCI and neurites that label for ubiquitin and p62 but not TDP-43, suggesting that some protein in addition to TDP-43 may be accumulating abnormally. Finally, the chromosomal region linked to disease phenotype in VSM-20 is particularly informative when compared with other 9p-linked FTD-ALS kindreds.
Clinical and neuroimaging features of VSM-20
Whereas the proportion of individuals in this family with each phenotype was similar,15 the mean age of onset (45.7 years) was earlier than in previously reported families.10 14 15 This was particularly apparent in the two pure ALS cases that had a mean onset of 37.5 years as compared with the cases with dementia that had a mean age of onset of 47.3 years. We speculate that VSM-20 individuals with dementia may have a less aggressive form of disease due to other genetic or environmental factors that mitigate the effects of the disease-causing mutation.
Severe limb apraxia, cortical sensory loss and Parkinsonism consistent with CBS23 has not previously been described in chromosome 9p linked FTD-ALS, although oral apraxia was reported in the family described by Morita et al.11 Of the VSM-20 ALS cases, all were limb onset, and Parkinsonism (mainly elevated tone and gait instability) was also seen in four family members. Interestingly, the neuroimaging analysis identified cortical white matter loss in VSM-20, but minimal basal ganglia volume loss as compared with the sporadic bvFTD group (table 2). These findings suggest that the elevated tone and hyper-reflexia in this family might be related to white-matter pathology and are consistent with the neuropathological analysis which demonstrated frontal subcortical and corticospinal tract white matter pathology in three VSM-20 cases. Similar patterns of grey and white matter reduction have previously been described in sporadic CBS cases,19 which suggests that the genetic abnormality in VSM-20 may be an unusual cause of this clinical syndrome. Overall, volume loss was most prominent bilaterally in the frontal lobes, with minimal temporal lobe atrophy in VSM-20. A similar pattern of atrophy has been associated with FTLD-TDP pathology in the presence of motor neuron disease in a previous study.5 A limitation of the MRI analysis was the relatively small number of VSM-20 subjects that were analysed and their clinical heterogeneity. It will be of interest to investigate patterns of brain atrophy in other 9p-linked families.
Neuropathological findings
In this report, we further provide the most detailed description of the neuropathology associated with 9p-linked familial FTD-ALS published to date. Consistent with previous reports, all three of our affected family members showed a combination of TDP-43-positive FTLD-U (FTLD-TDP) and classical ALS.10–12 14 15 The pattern of FTLD-TDP in all patients corresponded with FTLD-U type 37 (or type 2 according to the Sampathu, et al. classification). Although not numerous, TDP-43-ir GCI were also a consistent feature, suggesting a possible role of glia in the pathobiology of this condition.
There was some correlation between the clinical phenotype and the neuropathological findings in the three individuals who underwent autopsy examination. Case III-7, who had the most aggressive disease course, as indicated by the earliest age of onset and shortest duration, also had the most severe overall pathology. Although both III-7 and II-8 had pathological involvement of the pyramidal motor system, this was more extensive in III-7 who had clinical ALS compared with II-8 who did not. Finally, case III-7 who presented with CBS clinically had more severe pathological involvement of the striatonigral system. Consistent with the neuroimaging analysis which identified occipital lobe atrophy in VSM-20, we found evidence of occipital lobe pathology in cases III-7 and II-8 (supplemental table S3).
A consistent finding in our cases was several types of neuronal inclusions that were demonstrated with IHC for ubiquitin and p62 but not TDP-43. These have not been described previously in 9p-linked families and suggest the intriguing possibility that some protein other than TDP-43 might also be accumulating in specific neurons, perhaps the protein product of the mutant gene.
Refined localisation of the chromosome 9p FTD-ALS associated region
Genome-wide linkage analysis conclusively linked family VSM-20 to a 28.3 cM region between D9S1808 and D9S251 on chromosome 9p. Although a number of other FTD-ALS families have been linked to a similar chromosome 9p region, only one other single pedigree has previously generated a two-point LOD-score greater than 3.14 The minimum candidate region in VSM-20 overlaps with previously reported regions linked to FTD-ALS on chromosome 9p and significantly reduces the candidate region to a 3.7 Mb interval between D9S169 and D9S251, containing only 10 known or predicted genes (figure 5). However, consistent with previously published studies, extensive sequencing analysis of all exons and exon–intron boundaries in these 10 genes did not identify a disease-causing mutation in family VSM-20. Furthermore, real-time PCR analysis of the five protein coding genes in the region and analysis of genome-wide human SNP arrays in three patients of family VSM-20 failed to identify copy-number mutations. Together, these findings suggest that the FTD-ALS disease phenotype in family VSM-20 is caused by a mutation in a currently unassigned gene in the chromosome 9p region or by a small copy-number mutation, which could not be detected using the methods described. Although unlikely, it also remains possible that multiple FTD-ALS disease genes reside on chromosome 9p, reminiscent of FTD mutations in GRN and MAPT on chromosome 17q21,24 and that the disease gene in our family could be outside the region shared by all families.
Interestingly, the 3.7 Mb candidate region for familial FTD-ALS identified in this study overlaps with a novel susceptibility locus for sporadic ALS that was recently identified in a large-scale genome-wide association study.25 In that study, the strongest association was identified with an 80 kb haplotype block containing MOBKL2B, IFNK and c9orf72, three of the 10 genes also examined in this study.
In summary, we describe clinical, neuroimaging and neuropathological features of a novel FTD-ALS family with conclusive linkage to chromosome 9p. Affected members of this family displayed significant clinical heterogeneity. Similar variability has been reported in other chromosome 9p linked FTD-ALS families10–15 and is seen in all other autosomal dominant forms of FTD (including those caused by mutations in GRN, MAPT and VCP), indicating that other genetic or environmental factors must have a strong influence on the clinical phenotype.26–28 In addition to various combinations of bvFTD and ALS, Parkinsonism was noted in four family members and one individual initially presenting with apraxia and a corticobasal syndrome. White matter and parietal-occipital volume loss was more prominent in VSM-20, but temporal lobe volume loss was less apparent than in severity-matched sporadic bvFTD cases. All three family members examined neuropathologically displayed FTLD-TDP pathology with subtle differences in anatomical distribution that reflected the clinical and neuroimaging features. When combined with data from previous studies of other 9p-linked FTD-ALS families, this family's linkage results refine the candidate region from a 7.7 Mb to a 3.7 Mb interval. Future analysis of this minimal candidate region, conceivably by targeted next-generation sequencing, may ultimately reveal the mutational mechanism associated with chromosome 9p-linked FTD-ALS and the disease-causing mutation in family VSM-20.
Acknowledgments
We thank J Parisi and D Dickson, for providing neuropathological samples, and M Luk, J Gass, A Cannon, N Finch and M DeJesus-Hernandez, for technical assistance.
References
Supplementary materials
Web Only Data jnnp.2009.204081
Files in this Data Supplement:
Footnotes
Funding This work was supported by the National Institutes of Health (K23NS48855 to ALB, R01AG031278 to ALB, P01AG019724 to BLM, P50AG0300601 to BLM, P50AG16574 to BB and RR and R01 NS065782 to RR); the John Douglas French Foundation (ALB); the Hellman Family Foundation (ALB); the L. Hillblom Foundation (BLM); the ALS association (RR); the Canadian Institutes of Health Research (IM, HF); the Pacific Alzheimer Research Foundation (IM, HF and RR); and the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program of the Mayo Foundation (BB).
Competing interests None.
Patient consent Obtained.
Ethics approval Ethics approval was provided by the institutional review boards and ethics committees at the Mayo Clinic, University of British Columbia (UBC) and University of California, San Francisco (UCSF).
Provenance and peer review Not commissioned; externally peer reviewed.