Article Text

Abstract
Background Despite considerable interest, the population-based frequency, clinical characteristics and natural history of cognitive impairment in amyotrophic lateral sclerosis (ALS) are not known.
Methodology The authors undertook a prospective population-based study of cognitive function in 160 incident Irish ALS patients and 110 matched controls. Home-based visits were conducted to collect demographic and neuropsychological data. Patients were classified using the recently published consensus criteria and by a domain-based classification of both executive and non-executive cognitive processes.
Results 13.8% of patients fulfilled the Neary criteria for frontotemporal dementia. In addition, 34.1% of ALS patients without evidence of dementia fulfilled the recently published consensus criteria for cognitive impairment. Non-demented ALS patients had a significantly higher frequency of impairment in language and memory domains compared to healthy controls. These deficits occurred primarily in patients with executive dysfunction. 14% of ALS patients had evidence of cognitive impairment without executive dysfunction, and no cognitive abnormality was detected in almost half the cohort (46.9%).
Conclusion Co-morbid dementia occurs in approximately 14% of patients with a new diagnosis of ALS. Cognitive impairment, predominantly but not exclusively in the form executive dysfunction, is present in more than 40% of ALS patients who have no evidence of dementia. Cognitive impairment in ALS is not a universal feature, and its manifestations may be more heterogeneous than previously recognised.
- ALS
- motor neuron disease
- dementia
- cognition
- epidemiology
Statistics from Altmetric.com
Introduction
Amyotrophic lateral sclerosis (ALS) is primarily a motor system degeneration. However, it is now well recognised that there is clinical, pathological and genetic overlap between ALS and frontotemporal dementia.1 2 Previous studies have suggested that a subgroup of patients with ALS meet the criteria for the frontotemporal dementia (ALS-FTD), while other patients may display evidence of milder deficits in cognitive function and behaviour.3 4
From a clinical perspective, cognitive impairment in ALS has practical implications for patient management and prognosis.5–7 However, the range and frequency of cognitive impairment in ALS has not been fully characterised. This is primarily a function of the non-standardised methodologies utilised in many studies and because most studies to date have been drawn from small and clinic-based cohorts.1 5
Although preliminary consensus criteria have been generated in an attempt to define the cognitive and behavioural frontotemporal syndromes in ALS,5 these have yet to be validated. There remains an urgent requirement to fully characterise the frequency and range of cognitive impairment in ALS in a population-based setting. This in turn will help to provide a robust classification system that can be fully validated and incorporated into the existing El Escorial Criteria for ALS.
Using the Irish ALS register, we have conducted a population-based study of cognitive impairment in ALS using age-matched, sex-matched and education-matched controls. Our objective was to define the true extent and pattern of cognitive impairment in non-demented ALS patients examined within one year of diagnosis.
Materials and methods
Population-based recruitment was achieved through the Irish ALS Register, which has been in operation since 1995 and has been previously described.8 9 All recruited patients met the criteria for possible, probable or definite ALS as defined by the El Escorial diagnostic criteria for ALS.10 Exclusion criteria included history of other neurologic conditions that could affect cognition (major hemispheric stroke, traumatic brain injury, learning disability and severe active epilepsy), alcohol dependence syndrome, severe active mental illness and use of high-dose psychoactive medication.
Home-based semistructured interviews were conducted to collect the demographic and clinical data. Disease severity was assessed using the revised ALS Functional Rating Scale (ALSFRS-R).11 Arterialised capillary blood gas tensions were documented using a mobile transcutaneous sensor (TOSCA 500, Radiometer Ireland Ltd, Dublin). A detailed neuropsychological assessment lasting approximately 2 h was performed during these home visits.
The neuropsychological evaluations
The neuropsychological battery employed a range of standardised neuropsychological instruments (table 1).12–18 The design of the battery was based on the available literature which suggests early involvement of frontally mediated executive functions in ALS patients. The battery also screened for impairment in non-executive cognitive domains, in particular memory function. However, it is recognised that cognitive tests are rarely task-pure and in many instances can examine multiple cognitive processes. For example, executive function contributes to performance in some of our non-executive tasks, particularly the Rey–Osterrieth Complex Figure Test and memory tasks assessing encoding and retrieval.19 20 Similarly, perceptual processes are closely involved with accurate copy and reproduction of the Rey–Osterrieth Complex Figure Test. The majority of tasks were not timed, and adjustments were made for bulbar/motor disability in tasks that were time dependent (table 1).
Tasks included in the neuropsychological battery classified by cognitive domain and the definitions used for dysfunction in each cognitive domain
Premorbid Full-Scale IQ (FSIQ) was estimated using Wechsler Test of Adult Reading.21 The Pyramid and Palm Trees Test, a test of associative semantic knowledge, was used in further evaluation of patients in whom semantic language difficulties were suspected. This test has a number of potential versions, and in the current study, the non-verbal picture version was utilised.22 Mood was assessed using the Hospital Anxiety and Depression Scale.23 One item “I feel slowed down” was removed as it was felt it may be confounded by the limitations of physical disability in ALS patients.
Age-matched, sex-matched and education-matched healthy controls underwent the same neuropsychological assessment. The controls were recruited through a network that included the patients' spouses and primary care providers. The same exclusion criteria as for ALS patients were applied.
Patient categorisation
Co-morbid frontotemporal dementia was defined as a behavioural or language disorder that fulfilled the Neary criteria for frontotemporal dementia.24 Information on behaviour was obtained by direct evaluation of the patients and via semistructured interviews with the carers.
Diagnosis of other co-morbid dementias was reliant on the criteria recommended by Diagnostic and Statistical Manual of Mental Disorders-IV criteria (DSM-IV, American Psychiatric Association 1994) and the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association (NINCDS-ADRDA).25
ALS patients in whom the diagnosis of dementia was excluded were further stratified based on their neuropsychological performance. This cohort was first classified using the recently published consensus criteria.5 Cognitive impairment was defined as impairment on two tests of executive function that was below the fifth percentile of healthy controls.
The patients were also classified using a specifically designed cognitive domain-based classification. This method differed from the consensus criteria in two ways: a more stringent cut-off of two standard deviations below the mean for healthy controls (2.3rd percentile as opposed to 5th percentile) was used to define abnormal performance on any task; second, the presence of impairment in non-executive cognitive domains was taken into account, in addition to the number of cognitive domains involved.
Case ascertainment, clinical diagnosis and categorisation were supervised by a senior ALS specialist (OH) and a senior neuropsychologist (NP).
Statistical methods
The initial section of our results aims to describe the frequency of dementia and cognitive impairment in our cohort and the demographic profile associated with these syndromes. Following detailed exploratory analysis, means/medians were used for continuous variables and proportions for categorical variables. Comparisons were made using χ² test for categorical variables and two-sample t test or one-way ANOVA for continuous variables. Where data failed to meet criteria for parametric analysis, they were either transformed according to statistical standards, or non-parametric methods were used.
The second section of the results aims (1) to explore the sensitivity of the different executive tasks to cognitive impairment in our cohort and (2) to confirm that the differences in neuropsychological performance among the different patient subgroups were not due to differences in baseline demographics. We examined in detail the patients' performance on five tasks of attention/executive function (written phonemic verbal fluency index (wVFI)), category fluency, the Stroop Colour Word task, backward digit span task and Brixton Spatial Anticipation Test) and four tasks assessing different aspects of memory function (Logical Memory immediate story recall, Logical Memory delayed story recall, Logical Memory story retention and the Auditory Delayed Recognition Task). Multiple one-way ANCOVAs were undertaken using three covariates: age at time of assessment, education (in years) and disease severity at baseline (ALSFRS-R score). Bonferroni adjusted α value was p=0.006.
In the last section, the performance of ALS patients in whom no abnormality was detected was compared to that of healthy controls on all tasks in the neuropsychological battery (table 1). Bonferroni adjusted α value was set at p=0.002.
All tests were two tailed. Statistical significance was set at p<0.05. Bonferroni correction method was applied to adjust the α value in case of post hoc analysis and where multiple comparisons were undertaken. Statistical analyses were carried out using SPSS V.17 (SPSS Inc).
Results
Selection of participants
In the period extending from 1st of November 2006 to 30th of September 2010, 342 ALS patients were identified and included on the Irish ALS register. Of these, 207 patients were recruited to the study (60.5%, figure 1). Twenty-seven patients were subsequently excluded for the following reasons: history of major hemispheric stroke (n=7), alcohol dependency syndrome (n=5), active major psychiatric illness (n=3), severe seizure disorder (n=1), premorbid learning disability (n=1), presentation and course suggesting an atypical variant (n=4) and six patients were deemed too unwell to participate at the time of the home visit. A further 20 patients were deemed ineligible because the time between diagnosis and assessment exceeded 12 months, rendering them prevalent rather than incident cases.
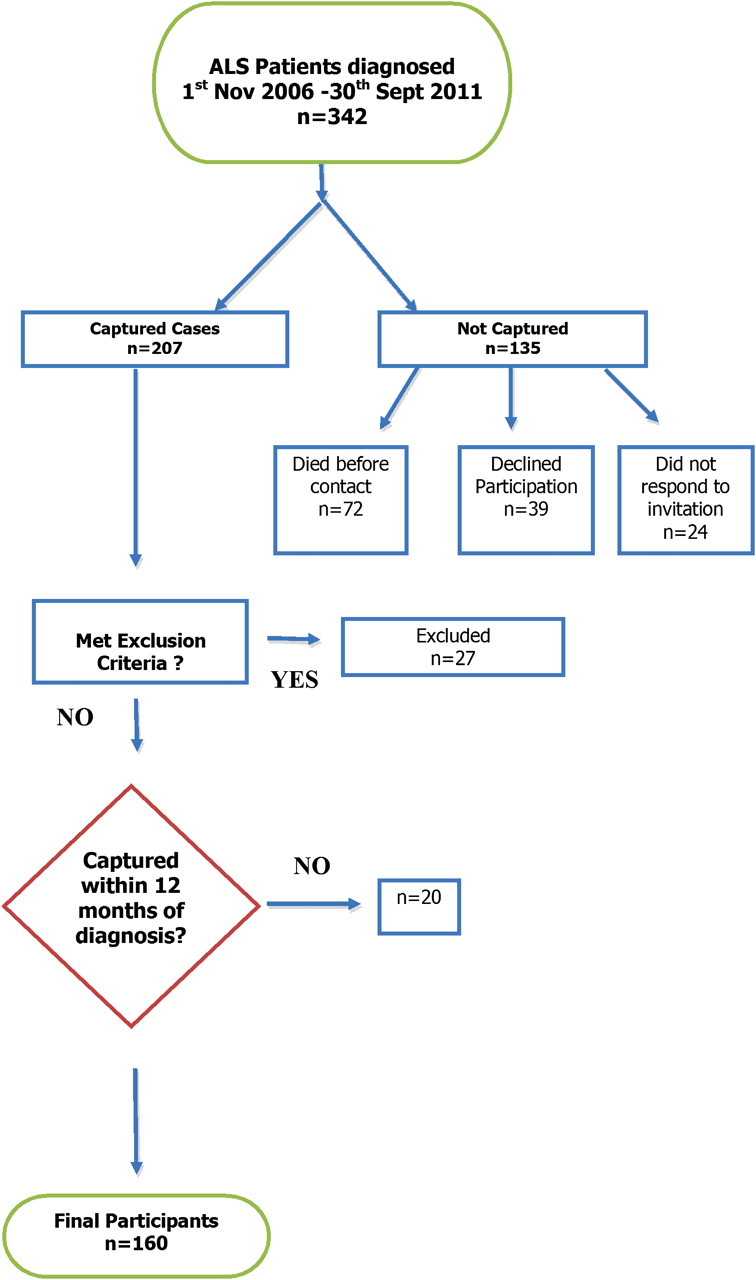
Flowchart showing capture rate and the sequence of participant selection. ALS, amyotrophic lateral sclerosis.
The demographics of the 160 incident patients included in the final analysis were compared with non-participants (including the excluded patients, n=182). Non-participants had a significantly higher mean age at diagnosis compared to participants (68.1 vs 63.1 years, p<0.0001). However, there was no significant difference between the two groups in gender distribution (p=0.422), bulbar onset (p=0.617) or delay to diagnosis (p=0.297).
Baseline characteristics of the participants (n=160)
The mean age of the participants at time of assessment was 63.5 years (range 34.7–87.3 years), and 61.5% were men. Fifty-five patients (34.2%) had bulbar-onset ALS, three patients (1.9%) had respiratory-onset ALS, while the remaining patients (n=103, 64.0%) had spinal onset disease.
Median time from symptom onset to diagnosis was 10 months. Median ALSFRS-R score at baseline was 38 (range 12–48). At the time of assessment, 137 patients (85.1%) were taking Riluzole, 21 patients (13.0%) had an enteral tube in situ and 14 patients (8.7%) were using non-invasive ventilation. Blood gases were documented using a transcutaneous sensor at time of assessment in 152 patients (94.4%). None of the patients had evidence of hypoxaemia (oxygen saturation <92 mm Hg).
Frontotemporal and co-morbid dementia
Three patients (1.9%) had evidence of co-morbid Alzheimer's type dementia. Twenty-two ALS patients (13.8%) fulfilled the Neary criteria for frontotemporal dementia (ALS-FTD).24 There were no significant differences between ALS-FTD patients and ALS patients without dementia (n=135) in age at symptom onset, gender, site of onset, time from symptom onset to assessment, FSIQ or number of years of formal education.
The majority of ALS-FTD patients (17/22) presented with behavioural changes typical of behavioural variant FTLD (bv-FTD). Two patients presented with expressive language difficulties typical of Progressive Non-Fluent Aphasia, and the clinical presentation in three patients was consistent with Semantic Dementia.
Cognitive status of ALS patients without dementia
One hundred and thirty-five patients (84.4%) had no evidence of dementia according to DSM-IV, NINCDS-ADRDA or the Neary criteria.24 25 Of these, three patients undertook fewer than two tests of executive function and thus could not be further categorised. The baseline characteristics of the remaining cohort (n=132) and those of 110 age-matched and sex-matched healthy controls are shown in table 2.
Baseline demographics of ALS patients without dementia (n=132) compared to those of healthy controls (n=110)
The consensus criteria
According to the consensus criteria,5 45 patients were cognitively impaired representing 34.1% of ALS patients without dementia (or 28.1% of the entire cohort). This rate was significantly higher than that of healthy controls (n=7, 6.4% p<0.0001).
The domain-based classification
The criteria used to define dysfunction in each cognitive domain are listed in table 1. Compared to healthy controls, non-demented ALS patients had significantly higher frequency of impairment in three out of four cognitive domains: executive dysfunction (25.8% vs 2.7%, p<0.0001); language dysfunction (23.3% vs 3.7%, p<0.0001); memory dysfunction (11.2% vs 1.8%, p=0.01) and visuo-spatial dysfunction (13.3% vs 5.5%, p=0.080).
Non-demented ALS patients were then divided into three groups based on the presence or absence of cognitive impairment and whether the executive domain was involved:
Impairment in executive dysfunction (ALS-Ex) single domain or in association with impairment in other cognitive domains (multidomain);
Non-executive cognitive impairment (ALS-NECI), single domain (eg, language only) or multidomain dysfunction (eg, language and memory);
No cognitive impairment.
The final classification of the whole cohort after using this domain-based categorisation is shown in figure 2.

{kind=link}
{kind=link}
This figure illustrates the final categorisation of the cohort of incident ALS patients (n=160) using the cognitive domain-based categorisation. Percentages were rounded to nearest whole per cent.ALS, amyotrophic lateral sclerosis; ALS-FTD, patients with ALS who meet the criteria for the frontotemporal dementia.
The frequency of each cognitive subgroup in non-demented ALS patients was compared to that in healthy controls (table 3). A significantly higher proportion of ALS patients had cognitive deficits characterised by executive dysfunction, particularly in combination with impairment of other cognitive domains. In the absence of executive impairment, the frequency of memory, language and/or visuo-spatial deficits was not significantly higher in ALS patients compared to controls.
Non-demented ALS patients (n=132) and age-matched, sex-matched and education-matched healthy controls (n=110) stratified using the proposed cognitive domain-based categorisation
Patients with executive dysfunction were older at symptom onset (p=0.011) and had more rapid disease progression (p=0.013, see table 4). Disease progression was estimated using decline in ALSFRS-R score since symptom onset (48 minus ALSFRS-R score divided by time from symptom onset to date of assessment). In addition, cognitively intact patients had significantly higher education and estimated premorbid FSIQ compared to the other two cognitive groups. A significant correlation was noted between education and FSIQ (Spearman's ρ 0.632, p<0.001).
This table shows the baseline characteristics of the three main cognitive categories of ALS patients without dementia
Cognitive performance and predictors of impairment
Phonemic verbal fluency index was identified as particularly sensitive task to executive impairment in non-demented ALS patients (see table 5; note that executive impairment was defined as abnormal performance on at least two tasks of executive function). The verbal fluency index task (written or spoken) was abnormal in 93.8% of patients with executive dysfunction, 30.0% of patients with non-executive cognitive impairment (NECI) and 9.7% of patients in whom no other cognitive abnormality was detected.
This table shows the comparison of the neuropsychological performance of ALS patients without dementia (ALS) and healthy controls (HC) on tasks of attention/executive function. This includes group mean scores and the proportion of each group with abnormal performance. Abnormal performance defined as a score that is two standard deviations below the mean for healthy controls. Results for written and spoken phonemic verbal fluency were merged for this analysis
After adjusting for age, disease severity and education, we investigated the performance of the three cognitive subgroups of ALS patients on selected tasks of executive and memory function (see methods section). There were significant intergroup differences in all tasks with the exception of the Brixton spatial anticipation test and backward digit span task (table 6).
Table shows mean scores of ALS cognitive groups on the selected executive and memory tasks and intergroup differences on multiple one-way ANCOVA
Post hoc analysis revealed that after adjusting for age, education and disease severity, patients with executive impairment had a significantly worse performance compared to two other subgroups on the wVFI, category fluency and the Stroop tasks (p≤0.002 in all comparisons). There was no significant difference between cognitively intact patients and patients with NECI on any of the executive tasks.
Patients with NECI performed poorly on the memory tasks, significantly worse than cognitively intact patients on all four memory measures (p<0.0001 in all comparisons). The performance of patients with executive dysfunction on the memory tests was more task-dependant. Compared to cognitively intact patients, they had significantly worse mean scores on the immediate and delayed story recall trials (p<0.0001 both cases), but not the story retention (p=0.010) or recognition task (p=0.064).
Cognitively intact patients
The mean scores of cognitively intact patients (n=75) were not significantly lower than those of healthy controls (n=110) on any of the tasks listed in table 1.
Discussion
This is the first large prospective population-based study of cognitive impairment in ALS. The demographics of our cohort were comparable to those of the incident cohorts in our previously published studies based on the Irish ALS register—a finding that is consistent with the population-based design of these studies.6 9 This study was based exclusively on incident ALS cases, and care was undertaken to identify age-matched, gender-matched and education-matched controls.
Co-morbid frontotemporal dementia was identified in 13.8% of incident ALS patients, a rate comparable to that reported by Rhingholz et al,3 but at variance with lower estimates. The majority of ALS-FTD patients presented with features consistent with bv-FTD. Our previous survival data suggested that ALS-FTD patients and patients with executive dysfunction have shorter survival time.6 This in turn suggests that studies reliant on prevalent cases are likely to underestimate the incidence of these frontotemporal syndromes.
In ALS patients without dementia, we observed an over-representation of executive dysfunction. The incidence of executive impairment in this cohort was higher using the consensus criteria (34.1%) compared to the domain-based criteria (25.8%). This is a direct reflection of the more stringent cut-off used by the domain-based criteria. Verbal fluency (phonemic and semantic) and the Stroop Colour Word task were identified as sensitive tasks to executive dysfunction in our cohort. This may be related to the heavy demands placed by these tasks on multiple frontostriatal circuits leading to sensitivity to any lesion affecting integrity of the prefrontal-circuit system.26 27
Our data also suggest that non-demented ALS patients have a higher frequency of language and memory dysfunction compared to healthy controls. Using the domain-based classification, we demonstrated that these deficits tended to occur in the context of executive dysfunction. This implies that in studies where patients with executive dysfunction are under-represented, the frequency of language, memory and visuo-spatial impairment in ALS may also be underestimated.
While language impairment in ALS is well described,28 29 the literature on memory function in ALS patients has been inconsistent, prompting our use of a range of memory tests. Reports of poor recall but intact recognition suggested that deficits may be due to abnormal retrieval or encoding processes, which are dependent on prefrontal function.15 30 However, deficits in delayed memory and recognition have also been reported.3 31 32
In our study, patients with NECI performed poorly on all memory tasks. Conversely, ALS patients with executive impairment performed poorly on immediate and delayed story recall, but not story retention or recognition. Executive processes subserved by a frontostriatal circuit are thought to contribute to encoding and retrieval processes in the first two tasks while the latter tasks, particularly recognition, are linked to the hippocampal memory system.20 33 This suggests that memory impairment in patients with executive dysfunction predominantly reflects impairment in the frontostriatal system while that in ALS-NECI extends to involve the medial-temporal memory system. These findings support the concept that cognitively impaired ALS patients may represent a heterogeneous population.
In our cohort, patients with executive dysfunction were older at symptom onset, had more rapidly progressive disease and had significantly lower education and premorbid IQ compared to those with intact cognition. Age, education and premorbid IQ are considered important predictors of cognitive reserve and function including processing speed and executive function.34 However, the executive deficits in this subgroup were still apparent after adjusting for these factors, suggesting that they are more likely to be markers for a particular subgroup of ALS that is more susceptible to an aggressive form of the disease. Cortical involvement in this group is predominated by early frontal involvement which, in some patients, extends to involve more posterior functions leading to multidomain involvement.
Multidomain executive impairment was present in 14.4% of non-demented ALS compared with 0% of healthy controls. Of the 19 ALS patients who had multidomain executive impairment, 15 had language impairment, 9 had visuo-spatial impairment and 6 had memory impairment. This suggests that the cognitive deficit in a considerable proportion of ALS patients with executive impairment may extend to more posterior domains. This will require further a more specialised cognitive investigation.
The frequency of NECI in ALS patients was not significantly higher than that in healthy controls. We cannot exclude the possibility that the NECI subgroup represents an incidental finding. It is also possible that our study was underpowered to detect a small but discrete subcohort of patients with a more ‘posterior’ form of cognitive impairment in ALS. Of note, one third of these patients had abnormal performance on the verbal fluency task. This may be a harbinger of more apparent frontal dysfunction that would emerge with time.
The heterogeneity of site of onset and the concept of focal onset followed by ‘linear spread’ to other cortical areas would be analogous to our current understanding of motor pathology in ALS.35 However, this hypothesis will require confirmation with longitudinal data and pathological input.
No significant difference was observed between the neuropsychological performance of cognitively intact ALS patients and that of healthy controls. Notwithstanding the presence of abnormal verbal fluency in 9.7% of these patients, our data suggest that at least 40% the ALS population appear cognitively intact.
This study is limited to an analysis of cognitive impairment. Subtle behavioural impairment not fulfilling the criteria for dementia was not included in the current analysis. Our neuropsychological battery was designed to minimise the effects of motor and physical disability. However, no adjustment was made for physical disability in category fluency. This may have resulted in lower scores in patients with bulbar dysfunction. In addition, as our primary focus was on executive and memory function, and due to limitations imposed by patient fatigue, language and visuo-spatial skills were screened using a single task for each domain. This may have reduced the scope for identifying more subtle deficits; thus, our findings in these cognitive domains require further, more detailed investigation.
In conclusion, our findings, drawn from a large population-based sample of ALS patients with appropriately matched controls, demonstrate that cognitive impairment may occur in more than 40% of incident patients with ALS. The deficits are primarily but not exclusively dysexecutive in nature, suggesting that ALS with cognitive impairment is a heterogenous group. Large studies using standardised criteria, and supported by radiological and pathological input, are required to confirm our observations. Further analysis incorporating behavioural change and documenting the evolution of the different cognitive syndromes over time is currently underway.
Acknowledgments
We would like to acknowledge all the patients who kindly agreed to participate in this research project.
References
Footnotes
JP and ME are joint first authors of this paper.
Funding The research leading to these results has received funding from the Health Seventh Framework Programme (FP7/2007-2013) under grant agreement n° 259867, ALSA (the ALS Association), HRB (the Health Research Board, grant H01300) and Research Motor Neuron (previously named Motor Neuron Disease Research Foundation).
Competing interests None.
Patient consent Obtained.
Ethical approval Written informed consent was obtained from all participants. The study had full ethical approval from Beaumont Hospital Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.