Article Text

Abstract
Background The aetiology and pathogenesis of non-genetic forms of frontotemporal dementia (FTD) is unknown and even with the genetic forms of FTD, pathogenesis remains elusive. Given the association between systemic inflammation and other neurodegenerative processes, links between autoimmunity and FTD need to be explored.
Objective To describe the prevalence of systemic autoimmune disease in semantic variant primary progressive aphasia (svPPA), a clinical cohort, and in progranulin (PGRN) mutation carriers compared with neurologically healthy normal controls (NC) and Alzheimer's disease (AD) as dementia controls.
Design Case control.
Setting Academic medical centres.
Participants 129 svPPA, 39 PGRN, 186 NC and 158 AD patients underwent chart review for autoimmune conditions. A large subset of svPPA, PGRN and NC cohorts underwent serum analysis for tumour necrosis factor α (TNF-α) levels.
Outcome measures χ2 Comparison of autoimmune prevalence and follow-up logistic regression.
Results There was a significantly increased risk of autoimmune disorders clustered around inflammatory arthritides, cutaneous disorders and gastrointestinal conditions in the svPPA and PGRN cohorts. Elevated TNF-α levels were observed in svPPA and PGRN compared with NC.
Conclusions svPPA and PGRN are associated with increased prevalence of specific and related autoimmune diseases compared with NC and AD. These findings suggest a unique pattern of systemic inflammation in svPPA and PGRN and open new research avenues for understanding and treating disorders associated with underlying transactive response DNA-binding protein 43 aggregation.
- DEMENTIA
- EPIDEMIOLOGY
- IMMUNOLOGY
- RHEUMATOLOGY
Statistics from Altmetric.com
Background
An inflammatory contribution to neurodegenerative disease pathogenesis has long been hypothesised.1 Alzheimer's disease (AD), frontotemporal dementia (FTD) and many other neurodegenerative conditions are united by pathological protein misfolding and aggregation accompanied by synaptic and neuronal loss and inflammatory markers around the site of pathological injury. Several studies have reported a lower prevalence of AD among those taking anti-inflammatory medications, suggesting a potential role for inflammation in AD.1 Nevertheless, it remains unclear whether inflammation plays a primary or secondary role in the major neurodegenerative conditions.
Frontotemporal lobar degeneration (FTLD) shows pathological abnormalities that are distinct from AD and thus provides an alternative disorder to investigate the relationship between inflammation and neurodegeneration. Previous studies of environmental risk factors in sporadic behavioural variant FTD found a significant association with head trauma and a close to significant association with thyroid disease, although that study lumped all of the FTD subtypes together without regard for neuropathological subsets.2 Furthermore, elevations in cerebrospinal fluid cytokines, notably tumour necrosis factor α (TNF-α), have previously been demonstrated in FTD.3 While provocative, these studies were performed before the full spectrum of FTLD pathological subtypes had been elucidated. Consequently, the patient population examined represented a heterogeneous mix of pathologies, predominantly FTLD due to τ aggregation and FTLD with abnormal cytoplasmic localisation of transactive response DNA-binding protein 43 (TDP-43) (FTLD-TDP). Therefore, it remains unclear whether systemic inflammatory illness was over-represented among patients with any clinical or pathological subtype.
In contrast to the heterogeneity of most of the FTD subtypes, semantic variant primary progressive aphasia (svPPA) is nearly always associated with underlying TDP-43 aggregates (75%–100% in clinicopathological correlation series).4 ,5 In our pathology confirmed cases at the University of California San Francisco (UCSF) Memory and Aging Center, 21/23 svPPA patients showed TDP-43 type C aggregates making this a clinical disorder with which the underlying neuropathology can be predicted. Furthermore, among FTD syndromes, svPPA is the least likely to be familial,6 making it an ideal disorder to study the prevalence of non-genetic factors, such as chronic inflammation. Another TDP-43 associated FTLD subtype, caused by to mutations in granulin (GRN) leading to a systemic deficiency in the progranulin (PGRN) protein, is associated with immune alterations.7 PGRN knockout mice develop inflammatory arthritis and PGRN has demonstrated antagonistic effects on TNF-α signalling.7 Recently, antibodies to PGRN have been demonstrated in patients with histories of particular autoimmune conditions, lowering systemic PGRN levels by half, similar to levels found in PGRN mutation carriers.8 ,9
As with neurodegenerative disease, autoimmune disease is increasingly correlating syndromic presentation with underlying pathomechanism. In some cases, autoimmune conditions that were considered unrelated now reveal networks that detail closer underlying genetic and pathological ties, the so-called ‘clusters’, while in others such links are not present.10–12 Given the associations between PGRN and inflammation, we hypothesised that, compared with normal controls (NC) and AD, the TDP-43-associated diseases (svPPA and PGRN mutation carriers) would display evidence of specific inflammatory signalling, as measured by an increased prevalence of particular clusters of autoimmune disorders and elevated TNF-α signalling.
Methods
Standard protocol approvals, registrations and patient consents
All subjects underwent informed consent to share their clinical data for research purposes. The study of patients’ clinical data was approved by the human research committee at UCSF and Mayo.
Participants
All participants underwent a thorough and standardised history and physical examination including the collection of past medical history. We retrospectively identified 94 svPPA patients from UCSF with complete records and whose clinical features conformed to revised consensus diagnostic criteria for svPPA.13 An additional 35 svPPA patients were contributed by Mayo Clinic Jacksonville (MCJ) all of whom met consensus diagnostic criteria for svPPA for a total cohort of 129 patients with svPPA. We identified 23 PGRN mutation carriers from UCSF and 16 from MCJ with complete records for a total of 39 PGRN patients. Patients were included in the PGRN group if they had a mutation in GRN,9 regardless of whether they were symptomatic, and all clinical phenotypes were included for symptomatic patients. Two of the PGRN patients also had been identified in our clinical svPPA cohort.
Age, gender and education-matched NC subjects were selected from a larger set recruited into a study of normal ageing. Subjects were included into the healthy ageing cohort if they had a normal neurological exam, MRI scans without clinically evident strokes and were without cognitive deficits or diagnosis of major psychiatric disease. With the exception of untreated multiple sclerosis, past history of autoimmune disease was not exclusionary for the NC subject group. Subjects were consecutively chosen from those most recently enrolled, and any with incomplete medical history were excluded. With the addition of 60 subjects from MCJ, a total of 186 older healthy controls were included in the study.
We obtained age, gender and education-matched AD subjects who met National Institute of Neurological and Communicative Diseases and Stroke/Alzheimer's Disease and Related Disorders Association) criteria for Alzheimer's disease (AD) (NINCDS-ADRDA) criteria.14 Any subjects with incomplete charts or diagnoses of comorbid Lewy Body and or vascular disease were excluded. In all, 35 additional AD subjects were contributed by MCJ leading to a total of 158 AD subjects.
Identification and classification of autoimmune conditions
UCSF and MCJ charts were reviewed in a retrospective manner by a rater blinded to neurological diagnosis, screening for any evidence of autoimmune disease. Using the same established criteria at both sites,15 we searched medical records for evidence of individual autoimmune conditions and modified the criteria by removing motor neurone disease and including only type 1, but not type 2, diabetes mellitus as autoimmune conditions. Furthermore, we added chronic lymphocytic colitis, lichen sclerosis and vitiligo for which there is evidence of autoimmune aetiology16–18 to Rugbjerg's criteria after having encountered these conditions in the medical records (table 1). The physicians’ notes in the review charts represented data that spanned over a decade in many cases and employed the standard thorough history taking typical of a behavioural neurology encounter. Only notes with reference of past medical history were included.
Screen of autoimmune conditions
Determination of TNF-α concentrations in plasma
Because PGRN has been shown to have antagonistic effects on TNF-α signalling, we attempted to obtain more direct evidence of TNF-α mediation in subjects for whom these data were available. TNF-α concentration in frozen-EDTA plasma samples were measured in a subset of patients with svPPA (n=26), PGRN (n=24), and healthy controls (n=37) was determined by use of a commercial ELISA, the Human TNF-α Ultra-Sensitive Plate (Meso Scale Discovery). The lower limit of detection was 0.036 pg/ml and lower limit of quantification 0.6 pg/ml.
Statistical analysis
Analysis of variance was used to test for significance for continuous variables such as age, education, Mini Mental State Examination (MMSE) score, Clinical Dementia Rating (CDR) Total score and CDR Sum of Boxes score across diagnostic groups. For categorical variables such as gender and ethnicity, χ2 tests were used. Prevalence and comparison of autoimmune disease among the diagnostic groups were assessed for statistical significance using χ2 tests. In order to determine whether non-thyroid autoimmune conditions were predictive of diagnosis, we conducted follow-up hierarchical bivariate logistic regressions in which the dependent variable was a dichotomous diagnostic variable. In step 1, we entered nuisance covariates including age, gender and education. In step 2, we entered presence of thyroid disease, and in step 3, we entered our primary independent variable of interest, presence of non-thyroid disease. This approach enabled us to examine whether the presence of a non-thyroid condition was a significant predictor of diagnostic status after accounting for other demographic factors and even thyroid disease. ORs for the non-thyroid autoimmune conditions among the diagnostic groups were also computed. The above analyses were performed using SPSS V.20.0 (IBM Corp, Armonk, New York, USA). A t test was employed to compare TNF-α levels in the svPPA and PGRN versus NC cohorts.
Results
Study 1: svPPA versus NC versus AD
The patient groups were matched for gender, education and race. The control group had a significantly higher MMSE at presentation and significantly lower CDR Total and Sum of Boxes scores, as expected (table 2).
Population demographics
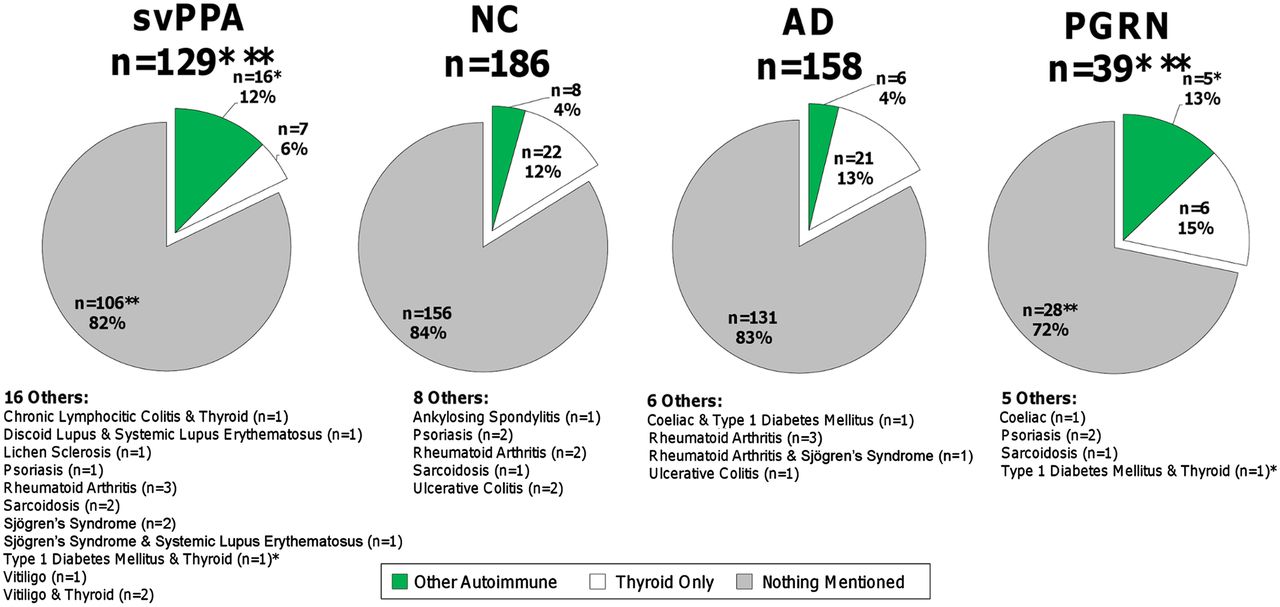
In the svPPA cohort, 18% (n=23) were positive for past medical history of autoimmune disease. Thyroid disease was the largest contribution to autoimmune disease in the svPPA cohort, 9% (n=11) in total. Of this group, 6% (n=7) had only thyroid disease. The four individuals with thyroid disease and another autoimmune condition had diagnoses of chronic lymphocytic colitis (n=1), type 1 diabetes mellitus (n=1) and vitiligo (n=2). The remaining 9% (n=12) presented with non-thyroid autoimmune disorders included: discoid lupus and systemic lupus erythematosus (n=1), lichen sclerosis (n=1), psoriasis (n=1), rheumatoid arthritis (n=3), sarcoidosis (n=2), Sjögren's syndrome (n=2), Sjögren's syndrome and systemic lupus erythematosus (n=1), and vitiligo (n=1). Analysis of the non-thyroid autoimmune conditions revealed significantly greater prevalence of disease in the svPPA cohorts compared with NC and AD (p=0.004) on χ2 analysis. In contrast, there were no significant differences in total autoimmune disease among the svPPA, NC and AD cohorts. Analysis of thyroid disease revealed no statistical differences in the prevalence of this condition across all groups and at rates comparable with general population estimates (8.9%–10.3%).19 ,20
Separate logistic regressions found that the presence of non-thyroid autoimmune disease predicted svPPA status above and beyond other variables for svPPA versus NC (OR=3.15, 95% CI 1.31 to 7.6) and svPPA versus AD (OR=3.59, 95% CI 1.36 to 9.46) (figure 1). No other variables in the model, including the presence of thyroid disease, were significant predictors of diagnostic status in either regression analysis.

Prevalence of autoimmune disease among diagnostic groups. AD, Alzheimer's disease; NC, normal controls; PGRN, progranulin; svPPA, semantic variant primary progressive aphasia. Retrospective chart review of autoimmune conditions in AD, NC, PGRN and svPPA subjects. Other Autoimmune refers to all other autoimmune conditions on the collection instrument that were not thyroid disease. When an individual had a thyroid disorder and another autoimmune disease, they were assigned to the Other Autoimmune category so as to avoid being counted twice. Thyroid Only refers to those who had only thyroid spectrum disorders affecting subjects. Nothing Mentioned refers to individuals where there was no mention of any condition found within the screening collection instrument. * and **Two patients in the collection overlapped between the PGRN and svPPA cohort (one had type 1 diabetes and thyroid disease and the other had no mentioned autoimmune disorder).
Study 2: PGRN versus NC versus AD
Comparison of the PGRN cohort with the previously obtained NC and AD groups revealed that the PGRN cohort was significantly younger. The groups were no different for education, gender and race. As was the case above, both the PGRN and AD cohort had significantly lower MMSE and significantly higher CDR Total and Sum of Boxes at presentation (table 3).
Population demographics
In the PGRN cohort, 28% (n=11) were positive for past medical history of autoimmune disease. Thyroid disease was the largest contribution to autoimmune disease in the PGRN cohort (n=6, 15%). Five other individuals (13%) presented with autoimmune diseases including: coeliac disease (n=1), psoriasis (n=2), sarcoidosis (n=1) and type I diabetes mellitus with thyroid disease (n=1). As with the svPPA group, analysis of the non-thyroid autoimmune conditions revealed significantly higher rates of disease in the PGRN cohorts compared with NC and AD (p=0.0002) on χ2 analysis. There were no significant differences in total autoimmune disease among the PGRN, NC and AD cohorts. Subgroup analysis of thyroid disease revealed no statistical differences in the prevalence of this condition across all groups and at rates comparable with general population estimates of thyroid disease (8.9%–10.3%).19 ,20
Bivariate logistic regression found that the presence of non-thyroid autoimmune disease predicted PGRN status above and beyond other variables for PGRN versus NC (OR=3.27, 95% CI 1.009 to 10.6) and PGRN versus AD (OR=3.73, 95% CI 1.1 to 12.9) (figure 1). No other variables in the model, including the presence of thyroid disease, were significant predictors of diagnostic status in either regression analysis.
PGRN and svPPA
Two PGRN patients in this collection also had clinical diagnoses of svPPA; this included an individual without any history of autoimmune disease and an individual with type I diabetes mellitus and thyroid disease. Both groups possess significant overlap with the types of rheumatological conditions, thyroid disease, inflammatory arthritides, cutaneous conditions and gastrointestinal disorders. Combining the PGRN and svPPA cohorts reveals that nearly all the non-thyroid conditions occur at higher rates than estimated rates in the general population (table 4).
Comparison of autoimmune prevalence
TNF-α analysis
Plasma concentrations of TNF-α were determined in all subjects for whom samples were available (PGRN, n=24, 62%; svPPA, n=26, 22.4%; NC, n=37, 21.5%). Compared with NC, plasma TNF-α concentrations were elevated in the PGRN and svPPA cohort. TNF-α for NC was 4.1±1.7 versus 6.0±3.8 pg/ml in PGRN (p=0.0075, t test) and versus 8.4±11 pg/ml in svPPA (p=0.012, t test) (figure 2).
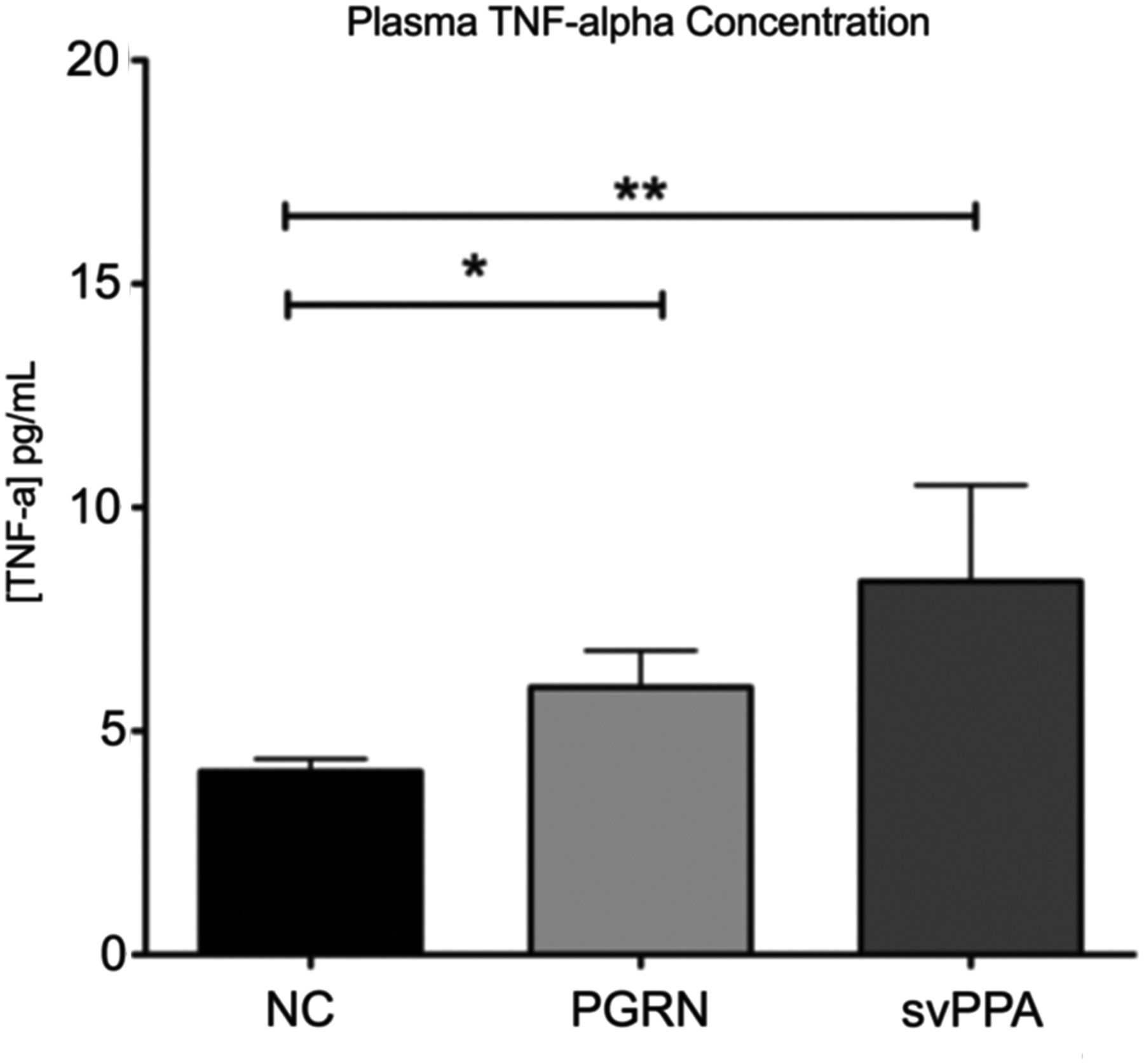
TNF-α levels in NC, PGRN and svPPA. NC, normal control; PGRN, progranulin; svPPA, semantic variant primary progressive aphasia; TNF-α, tumour necrosis factor α. Samples from NC,37 PGRN24 and svPPA26 were analysed for plasma TNF-α levels. * and **Statistical differences in TNF-α levels were found between NC and PGRN as well as NC and svPPA (p=0.0075 and p=0.012, respectively; t test).
Discussion
We observed a higher prevalence of specific autoimmune conditions in svPPA and in PGRN carriers compared with matched cohorts. Both svPPA and PGRN patients showed elevated levels of TNF-α compared with controls. These findings suggest a strong relationship between inflammation signalling, immune-mediated illness, and two neurodegenerative conditions, svPPA and PGRN mutations.
Literature on autoimmune pathogenesis finds that diseases tend to cluster in groups, although these groups have not yet been consistently defined.10–12 These clusters are determined by epidemiological overlap, conditions co-occurring in the same individual or within first-degree relatives at rates higher than expected for the general population, and by shared genetic risk alleles. In addition to showing similarities in autoimmune pathogenesis within clusters, some studies have also begun to show significant differences between various clusters.30
Thyroid disease is considered an archetype autoimmune disease cluster with particularly strong overlap with pernicious anaemia and diabetes.10 An indepth meta-analysis of genome-wide-association studies by Sirota et al30 revealed an inverse relationship between thyroid disease and rheumatoid arthritis clusters, providing further justification for considering thyroid disease separately from the other immune conditions found in our cohorts. Thyroid disease is the most prevalent autoimmune condition in the general population16 occurring at rates of nearly 10% of an elderly population.19 ,20 In our study, thyroid disease was the single greatest contribution to autoimmune disease across all cohorts, at rates statistically similar to each other and rates found in the general population. This argues against a specific association between thyroid disease and either svPPA or PGRN. This finding contrasts with a near significant association of bvFTD with thyroid disease previously reported by others.2
Another autoimmune cluster, defined by inflammatory arthritides rheumatoid arthritis, systemic lupus erythematosus and psoriasis,10–12 appeared prominently in both svPPA and PGRN cohorts. There are well-documented convergences between Sjögren's syndrome and sarcoidosis with rheumatoid arthritis, systemic lupus erythematosus and psoriasis including highly significant associations with increased TNF-α signalling, an abnormality found in svPPA and PGRN carriers.11 ,31–33
Other clusters prominently appearing in both svPPA and PGRN cohorts, cutaneous and gastrointestinal, have been less well characterised in the literature. Supporting a cutaneous cluster are the co-occurrences of and common T cell activation pathogenesis shared among discoid lupus, lichen sclerosis, psoriasis and vitiligo.18 ,34 ,35 Supporting the existence of a gastrointenstinal cluster, chronic lymphocytic colitis shares genetic and pathological features with coeliac disease.17 Taken together, autoimmune disorders belonging to each of these non-thyroid clusters were found to have higher rates in the svPPA and PGRN cohorts than in NC or AD controls and occur at rates greater than general population estimates.
With regard to the relationship between autoimmune disease and PGRN, an analysis of PGRN knockout mice revealed a susceptibility to inflammatory arthritis and high levels of TNF-α.7 Although this association has yet to be established in human GRN mutation carriers, our data would appear to support this link. GRN mutations result in FTLD-TDP, type A neuropathology, and clinicopathological studies demonstrate that svPPA is most often associated with underlying FTLD-TDP, type C pathology.36 Both of these FTLD-TDP disorders appear to be linked by autoimmunity. Our observation of a related pattern of systemic inflammatory disorders between PGRN and svPPA suggests that FTLD-TDP, type C, might have similar pathomechanisms. Finding increased TNF-α levels in both our PGRN and svPPA cohort further strengthens this potential link as an effective magnification of TNF-α signalling was hypothesised as a probable mechanism of this rheumatological disease vulnerability in the PGRN knockout mice.
Last, a recent publication revealed the presence of anti-PGRN antibodies in around 40% of screened rheumatoid arthritis (16/44) and systemic lupus erythematosus patients (39/91). These antibodies had the direct effect of lowering plasma PGRN levels by about 50% compared with NC,8 mirroring the haploinsufficiency effects of PGRN mutations.9 The presence of anti-PGRN antibodies in autoimmune disease provides a direct mechanism of action for how sustained autoimmune pathology would precipitate FTLD-TDP disease and supports our findings of increased rates of these related autoimmune disorders in FTLD-TDP populations.
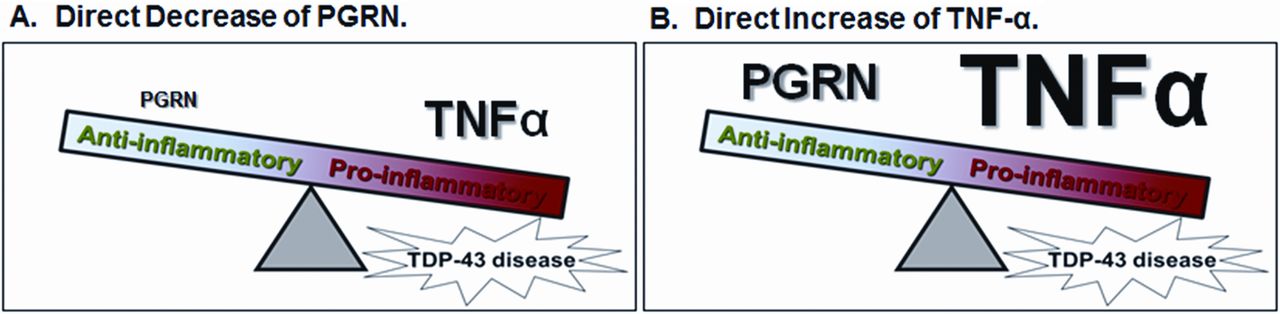
Based on the present work and previous studies, we propose a model in which an imbalance of anti- and pro-inflammatory factors results in systemic inflammation and susceptibility to specific neurodegenerative diseases (figure 3). In this model, increased TNF-α signalling, either via primary decreased PGRN expression (as seen in patients with GRN mutations or patients with autoimmune disease who develop anti-PGRN antibodies) and secondary increased TNF-α or primary increased TNF-α expression (which can occur in the setting of autoimmune disease as well as in chronic disease unrelated to autoimmune mechanisms), increases susceptibility to certain types of FTLD-TDP. These two mechanisms are not mutually exclusive and likely interact with each other. Currently, this model relates only to FTLD-TDP types A and C; however, it should be noted that the other well known FTLD-TDP-causing mutations, C9ORF72 and valosin containing protein (VCP), which are accompanied by FTLD-TDP types B (as well as A) and D, respectively,36 ,37 also have intriguing links to immune function, although these links require further study.
{kind=link}
{kind=link}
{kind=link}
Model of relative increased TNF-α signalling in TDP-43 disease, via PGRN depletion or TNF-α elevation. PGRN, progranulin; TDP-43, transactive response DNA-binding protein 43; TNF-α, tumour necrosis factor α. Proposed unifying schema for TDP-43 disease pathology mediated through the effects of TNF-α signalling. (A) Primary PGRN and secondary TNF-α mediated mechanisms; (B) A primary TNF-α mediated mechanism of TDP-43 disease. These two mechanisms likely interact with each other.
If confirmed, these findings may help delineate how specific patterns of systemic inflammation predispose to discrete forms of neurological disease. It will be exciting to see if, with larger numbers, patterns arise that allow for the prediction of specific underlying TDP-43 subtypes and whether the neurodegenerative disease will prove amenable to anti-inflammatory approaches. TDP-43 proteinopathy has become increasingly recognised as co-pathology in many neurodegenerative diseases, found in up to 50% of AD, 60% of Parkinson's disease and occasionally in patients with Huntington disease.38 As such, further study using PGRN and svPPA as model systems may help clarify TDP-43 pathobiology generally.
While this study has limitations—svPPA is a relatively uncommon disease and the cohort described here remains small despite our multi-centre approach; PGRN carriers are even rarer—our analysis represents among the largest collection of these patients to date and displays adequate power to detect significant increases in inflammatory disease prevalence in separate svPPA and PGRN cohorts. The collection of past medical history was performed in a retrospective manner based on previous physician diagnoses and obtained in open-ended questioning, rather than by direct laboratory-based evaluations of autoimmune markers. This suggests that we may have under-represented the prevalence of autoimmune disease in this sample. While all our patients receive the same attention regarding history and physical examination, NC and dementia patients alike, both UCSF and MCJ are tertiary care centres with specialty dementia care clinics. As such, it is possible that previous to visiting with our centres, the subjects in the PGRN and svPPA cohort may have received greater medical attention than the NC cohort. Nonetheless, our NC group showed a roughly similar prevalence of overall autoimmune disease to the other cohorts arguing against systematic ascertainment bias. The younger age of the PGRN cohort was driven by the inclusion of asymptomatic carriers. As rates of autoimmune disease increase with age, a younger experimental group with older controls would only bias against our hypothesis. TNF-α signalling was chosen as a marker of inflammation in an exploratory manner and in the future we hope to broaden the analysis of inflammatory markers to include additional cytokines, autoimmune antibodies and other measures of inflammation.
Despite these limitations, the present findings build on previous work39 ,40 and warrant careful review for a history of autoimmunity in all patients with neurodegenerative disease with particular emphasis on FTLD pathologies. These findings may open up a suite of new diagnostic tools and therapeutic approaches to FTLD-TDP. Whether systemic inflammation creates risk for TDP-43 disease or both autoimmune and TDP-43 disorders reflect shared underlying pathogenic mechanisms remains undetermined and should be pursued in future studies. Furthermore, we should entertain the possibility that autoimmunity may in some instances represent a preclinical disruption of neuroimmunological function.
Acknowledgments
We thank Dr Michael Weiss from the University of Washington for helpful discussions on the coincidence of neuromuscular and autoimmune disease.
References
Footnotes
-
Competing interests ZAM reports no disclosures. He was responsible for the conceptualisation and design of the study, collection, analysis, and interpretation of the data, drafting, and revising the manuscript. KPR is funded by NIH/NIA 1R01AG029577–01, P50AG023501 and Hillblom #2007/2I. She helped with the design and conceptualisation of the study, analysis and interpretation of the data, drafting, and revising the manuscript. NRG-R is funded by NIH grant P50AG16574 (Mayo ADRC Ronald Petersen PI). He is also funded by U01AG010483 from the ADCS and has funding for multi-centre studies from Allon, Pfizer, Janssen and Medivation. He contributed to the design and conceptualisation of the study, analysis and interpretation of the data, drafting, and revising the manuscript. LTT reports no disclosures. His contributions included analysis and interpretation of the data, and revising the manuscript. VES reports no disclosures. She helped with analysis and interpretation of the data and revising the manuscript. CMC reports no disclosures. Contributions included analysis and interpretation of the data, and revising the manuscript. LAC reports no disclosures. Her contributions included analysis and interpretation of the data, drafting and revising the manuscript. PAJ reports no disclosures. His contributions included design and conceptualisation of the study, analysis and interpretation of the data, drafting, and revising the manuscript. TS reports no disclosures. She contributed to the design and conceptualisation of the study, collection, analysis, and interpretation of the data, drafting, and revising the manuscript. KAH reports no disclosures. She helped with the collection, analysis and interpretation of the data, and revising the manuscript. AK reports no disclosures. Contributions included collection of data and revising the manuscript. BKK reports no disclosures. He contributed to analysis of the data and revising the manuscript. SCH reports no disclosures. She helped with collection and analysis of the data. LTG is funded by the John Douglas French Foundation and NIH grant R01AG040311-01. Her contributions included interpretation of the data and revising the manuscript. MLG-T reports no disclosures. Her contributions included data collection, drafting and revising the manuscript. ALB has been a consultant for Bristol Myers Squibb, Genentech, Plexikkon, Phloronol, Registrat-Mapi, Accera, Envivo, TauRx and Novartis, receives research support from Allon Therapeutics, Bristol Myers Squibb, Janssen, Forest, Pfizer, Medivation and Genentech, and is funded by NIH grants R01AG038791, R01AG031278, the John Douglas French Foundation, Alzheimer's Drug Discovery Foundation, the Association for Frontotemporal Degeneration, the Silicon Valley Foundation, the Agouron Institute, the Tau Research Consortium and the Hellman Family Foundation. His contributions included analysis and interpretation of the data and revising the manuscript. HJR is funded by NIH grant R01-AG032306 and has no disclosures. His contributions included data collection, drafting, and revising the manuscript. JHK has no disclosures. Contributions included drafting and revising the manuscript. GC has no conflicts of interest. He is funded by R01 AG026938. His contributions included analysis and interpretation of the data and revising the manuscript. DHG has no conflicts of interest. He is funded by the Alzheimer's Disease Research Center of California (ARCC) grant 03-7527 and R01AG026938. His contributions included analysis and interpretation of the data and revising the manuscript. RR is funded by NIH grants R01NS065782, R01AG026251, P50AG16574, the ALS Therapy Alliance, the Consortium for Frontotemporal Research, and has a patent pending on expanded non-coding repeat in C9ORF72 cause frontotemporal dementia and amyotrophic lateral sclerosis. Her contributions included analysis and interpretation of the data, revising the manuscript. WWS is funded by NIH grants P50 AG1657303, the John Douglas French Alzheimer's Disease Foundation, Consortium for Frontotemporal Dementia Research, James S McDonnell Foundation, Larry Hillblom Foundation, has received support for travel by the Alzheimer's Association, and received payment for lectures by the Alzheimer's Association, American Academy of Neurology and Novartis Korea. He helped with the design and conceptualisation of the study, analysis and interpretation of the data, drafting and revising the manuscript. TW-C is funded by NIH grants U01NS057496, R01AG030144 and the Department of Veterans Affairs. He helped with design and conceptualisation of the study, analysis and interpretation of the data, drafting, and revising the manuscript. BLM serves as board member on the John Douglas French Alzheimer's Foundation and Larry L Hillblom Foundation, serves as a consultant for TauRx, Allon Therapeutics, Siemens, BMS, the Tau Consortium and the Consortium for Frontotemporal research, has received institutional support from Novartis, and is funded by NIH grants P50AG023501, P01AG019724, P50 AG1657303 and the state of California. He contributed to the design and conceptualisation of the study, analysis and interpretation of the data, drafting, and revising the manuscript.
-
Funding This work was supported by National Institutes of Health (grants P01 AG19724, P50 AG023501, P50 AG1657303, P50AG16574, R01-AG032306 and R01 NS050915 -05A1) and its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute on Aging or NIH. Additional funds include the Consortium for Frontotemporal Dementia Research, the NSF Graduate Research Fellowship, the Tau Research Consortium and the Larry Hillblom Foundation grants 2002/2J and 2007/2I.
-
Competing interests None.
-
Ethics approval The study of patients’ clinical data was approved by the human research committee at UCSF and Mayo.
-
Provenance and peer review Not commissioned; externally peer reviewed.