Article Text

Abstract
Background Significant heterogeneity in clinical features of frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS) cases with the pathogenic C9orf72 expansion (C9P) have been described. To clarify this issue, we compared a large C9P cohort with carefully matched non-expansion (C9N) cases with a known or highly-suspected underlying TAR DNA-binding protein 43 (TDP-43) proteinopathy.
Methods A retrospective case-control study was carried out using available cross-sectional and longitudinal clinical and neuropsychological data, MRI voxel-based morphometry (VBM) and neuropathological assessment from 64 C9P cases (ALS=31, FTLD=33) and 79 C9N cases (ALS=36, FTLD=43).
Results C9P cases had an earlier age of onset (p=0.047) and, in the subset of patients who were deceased, an earlier age of death (p=0.014) than C9N. C9P had more rapid progression than C9N: C9P ALS cases had a shortened survival (2.6±0.3 years) compared to C9N ALS (3.8±0.4 years; log-rank λ2=4.183, p=0.041), and C9P FTLD showed a significantly greater annualised rate of decline in letter fluency (4.5±1.3 words/year) than C9N FTLD (1.4±0.8 words/year, p=0.023). VBM revealed greater atrophy in the right frontoinsular, thalamus, cerebellum and bilateral parietal regions for C9P FTLD relative to C9N FTLD, and regression analysis related verbal fluency scores to atrophy in frontal and parietal regions. Neuropathological analysis found greater neuronal loss in the mid-frontal cortex in C9P FTLD, and mid-frontal cortex TDP-43 inclusion severity correlated with poor letter fluency performance.
Conclusions C9P cases may have a shorter survival in ALS and more rapid rate of cognitive decline related to frontal and parietal disease in FTLD. C9orf72 genotyping may provide useful prognostic and diagnostic clinical information for patients with ALS and FTLD.
- Cognitive Neuropsychology
- Neurogenetics
- Frontal Lobe
- Mri
- Motor Neuron Disease
Statistics from Altmetric.com
Introduction
Over the past decade, significant evidence emerged supporting a clinicopathological continuum between amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD).1 This includes cognitive impairment in up to 50% of patients with ALS,2 subclinical motor neuron disease in FTLD,3 the presence of neuronal and glial inclusions composed of the RNA-binding protein TAR DNA-binding protein 43 (TDP-43) in both diseases,4 and families with members afflicted with one or both disorders linked to a locus on chromosome 9.5
The recently discovered hexanucleotide expansion in the C9orf72 gene appears to be the most common genetic cause of familial,6 ,7 ALS and FTLD, and is also found in a number of apparently sporadic cases.8 Due to the high prevalence of this mutation, information on the natural history and clinical features of this hereditary degenerative condition is very important for prognostic and diagnostic considerations. Previous reports find considerable heterogeneity in the clinical phenotype and demographic features of expansion-positive cases (C9P) as compared with non-autopsy-confirmed sporadic expansion-negative (C9N) cases.9–11 C9P cases have been shown to have an earlier age of onset,8 ,9 ,12–14 and to further assess the hypothesis that C9P cases may progress more rapidly, we evaluated longitudinal clinical data in C9P compared to C9N cases with known or highly-suspected TDP-43 neuropathology. Comparative studies using C9N FTLD cases with underlying TDP-43 neuropathology are essential to identify specific characteristics of this expansion, as care must be taken to minimise tauopathies (FTLD-tau) or atypical presentations of Alzheimer's disease (AD) that may contaminate a clinically-derived frontotemporal dementia (FTD) reference cohort.15
Methods
Patients
Patients were initially seen at the University of Pennsylvania Frontotemporal Degeneration Center (FTDC), ALS Center (ALSC), or AD Center (ADC). The clinical diagnosis of ALS was made using the El Escorial-revised criteria,16 and FTLD clinical phenotypes using established clinical criteria.17 ,18 Cognitive impairment in two patients with ALS not meeting criteria for FTD was diagnosed clinically as being non-amnestic mild cognitive impairment (ALS-MCI) and grouped with ALS cases for analyses below. Family history was assessed using a modified Goldman score.19 Briefly, score 1=autosomal dominant (three or more people in two generations with any combination of FTD or ALS with one person being a first-degree relative (FDR) of the other two), 2=familial aggregation (three or more relatives with dementia or ALS and criteria for autosomal dominant inheritance were not met), 3=a single affected FDR with dementia or ALS (age ≤65), 3.5=a single affected FDR with dementia or ALS (>65) and 4=no contributory family history or unknown family history. All procedures were performed in accordance with the Institutional Review Board at the University of Pennsylvania using an approved written consent procedure.
Genetic testing for the C9orf72 hexanucleotide repeat was performed in 222 patients (except 1 obligate carrier) with a clinical or neuropathological diagnosis of ALS and/or FTLD (67 C9P, 155 C9N).
After exclusion of cases with minimal clinical data and those likely to have neuropathologies other than TDP-43 in the C9N group,15 ,18 ,20–23 the final cohort consisted of 64 C9P cases and 79 C9N (figure 1). All C9P cases were unrelated except for two pairs. In the C9N group there were seven unrelated cases with known pathogenic progranulin (GRN) mutations,24 (GRN c.348A>C (p.Ser116Ser, predicted to cause splice site mutation), p.Arg110X, p.Arg418X, c.1179+2T>C (predicted to cause p.Val395TyrfsX29), p.Ser226TrpfsX28, p.Thr272SerfsX10, p.Asp441HisfsX4) known to have underlying TDP-43 neuropathology.23 Since our main outcome measures involved cognition and survival, patients were divided into two groups according to cognitive status: FTLD (FTLD/ALS-FTLD) and ALS without dementia (ALS; Table 1 ).

Flow chart of subjects included in analysis. CBS, corticobasal syndrome; lvPPA, logopenic variant PPA; naPPA, non-fluent/agrammatic variant PPA; PPA, primary progressive aphasia; PSP progressive supranuclear palsy.
Patient demographic information
Genetic analysis
DNA was extracted from peripheral blood or brain tissue and genotyped for the C9orf72 hexanucleotide repeat using a modified repeat-primed PCR,7 as previously described.25 GRN mutation testing of the cohort had been performed previously.24
Neuropsychological testing
Neuropsychological test scores were selected within 6 months of neuroimaging or, in the absence of neuroimaging, at the earliest testing visit. Longitudinal analysis included data from the most complete subsequent testing visit >6 months after baseline (median=13.8, range=6.2–43.4 months). Tests included the Mini-Mental State Examination (MMSE),26 and letter-guided verbal fluency. We focused on verbal fluency because of earlier reports demonstrating difficulty on executive measures in C9P.27 ,28 Verbal fluency scoring was determined by the total number of novel ‘F words’ generated in 1 min, with the exclusion of proper nouns and numbers. An annualised decline score was calculated by dividing the change in score between visits by the time interval (years) between evaluations. Additional cognitive measures available in smaller numbers of subjects are summarised in supplementary materials.
MRI acquisition and analysis
A subset of patients with FTLD (n=41) underwent a structural T1-weighted magnetisation-prepared rapid acquisition with gradient echo (MPRAGE) MRI acquired from a SIEMENS 3.0T Trio scanner with an eight-channel coil using the following parameters: repetition time=1620 ms; echo time=3 ms; slice thickness=1.0 mm; flip angle=15°; matrix=192×256; and in-plane resolution=0.9×0.9 mm. Whole-brain MRI volumes were preprocessed using PipeDream (https://sourceforge.net/projects/neuropipedream/) and Advanced Normalisation Tools (http://www.picsl.upenn.edu/ANTS/), as described,29 ,30 and resampled to 2 mm3 voxels. We analysed cortical thickness31 for the cerebrum. We performed a separate grey matter (GM) density analysis of the cerebellum because of the distinct anatomic characteristics of this region.
Analyses were performed using FSL's randomise module (http://www.fmrib.ox.ac.uk/fsl/randomise). Group differences were evaluated using an explicit mask to constrain voxel-wise comparisons to regions of GM and we used permutation-based (N=5000) methods for each test. For each patient group compared to healthy seniors, we report clusters that survive a p<0.05 (FDR-corrected) threshold and contain a minimum of 50 adjacent voxels. For direct comparison of C9P and C9N, we report clusters that survive a p<0.01 (uncorrected) threshold and contain a minimum of 50 adjacent voxels. We additionally performed a non-parametric linear regression analysis using FSL's randomise module to relate verbal fluency performance in imaging subjects to regions of significant GM disease. We constrained our analysis to regions of disease using an explicit mask generated from the direct comparisons of patient groups, since interpretation of a result in a region without disease would otherwise be difficult. For example, a significant brain-behaviour relationship in areas that did not differ between groups could be attributed to a variety of non-specific factors.
Neuropathological examination
Autopsy was performed on a subset of cases (n=64) using standard techniques as previously described.15 To evaluate the severity of neurodegeneration, haematoxylin and eosin (H&E)-stained slides were graded by trained examiners (DJI, JB) blinded to C9 status using a semiquantitative scoring system (0=none, 1=mild, 2=moderate and 3=severe) for neuronal loss and gliosis in four cortical regions, thalamus and granule layer of the cerebellum. A random sample (20% of regions) was re-rated independently by examiners (DJI, JBT) to evaluate inter-rater and intrarater reliability, and Spearman's correlations confirmed high reliability (r=0.84; p<0.001 and r=0.81; p<0.001, respectively). Ubiquilin (UBQLN and TDP-43) neuropathology was also examined using techniques as previously described.25
Statistical analysis
Between-group comparisons for clinical data were performed using independent-sample t tests, one-way and repeated-measure analysis of variance (ANOVA) tests and χ2 analyses, as appropriate. We report mean values±SE. Survival was calculated based on interval from the reported onset of symptoms to death and analysed using a Kaplan–Meier log-rank analysis. Non-normally distributed semiquantitative neuropathological scores and annualised neuropsychological decline scores were compared between groups using non-parametric Mann–Whitney U rank sum tests. Correlations between clinical and neuropathological variables were performed using Spearman correlations. We performed two-sided tests with a p<0.05 level of significance using SPSS V.19.0 (SPSS, Chicago, Illinois, USA).
Results
Clinical features and neuropsychological testing
The C9P group consisted of 64 patients (31 ALS, 33 FTLD) and the C9N group contained 79 patients (36 ALS, 43 FTLD) (figure 1). There were no significant differences in gender, percentages of clinical phenotypes, education and race between groups. There was a non-significant trend for a lower Goldman score (more substantial family history) in C9P than C9N (p=0.088). C9P cases had a significantly earlier age of onset (p=0.047), and in a subset of non-living cases, earlier death (p=0.014) compared to C9N (table 1). Subsequent analyses were performed separately on FTLD and non-demented ALS subgroups.
The majority of the C9P FTLD clinical phenotype was behavioural-variant FTD (bvFTD) (17/24), with one case of semantic variant primary progressive aphasia (svPPA) and four cases of non-fluent/agrammatic variant PPA (naPPA). Two C9P FTLD cases were clinically diagnosed as having probable AD,32 during life and had a neuropathological diagnosis of FTLD-TDP. The C9N FTLD group had a similar predominance of bvFTD (23/33). C9P FTLD cases had no significant difference in the age of onset, death or onset-death interval compared with the C9N FTLD group (table 1).
Neuropsychological data were available for 28 C9P group patients with FTLD and 41 C9N group patients with FTLD. Baseline testing was performed at approximately the same age and stage of illness relative to reported onset of symptoms for both groups (table 2). There was no significant difference between cross-sectional and longitudinally studied cases in race, education, gender, or clinical phenotype.
Neuropsychological test data in FTLD with and without the C9orf72 repeat expansion
C9P and C9N patients with FTLD had similar mean baseline MMSE (C9P=23.3, C9N=24.7) and verbal fluency (C9P=6.2, C9N=6.4) scores (table 2). A subset of these patients had follow-up testing data for MMSE (C9P=15, C9N=24) and verbal fluency (C9P=10, C9N=16), with similar time intervals between evaluations. There was a significantly greater decline in mean annualised letter fluency in C9P FTLD (4.5 words/year) compared to C9N FTLD (1.4 words/year; p=0.023). There was also a trend for a greater annualised decline in MMSE scores in C9P FTLD (p=0.078; table 2). Repeated-measure ANOVA tests found significant interaction of C9 status with MMSE (F(1,37)=5.2, p=0.028) and C9 status with verbal fluency (F(1,24)=6.6, p=0.017) scores between visits, suggesting more rapid decline on measures of global (MMSE) and executive-mediated (letter fluency) cognition for C9P FTLD compared with C9N. Individual case analysis found the majority of C9P FTLD with ≥3 word/year decline in letter fluency and ≥4 points/year in MMSE (see online supplementary table S1). Neither second visit MMSE or verbal fluency scores, nor annualised rates of decline for these tests correlated with the interval between testing. Additional neuropsychological testing showed confrontation naming and other executive impairments in C9P and C9N FTLD at baseline and at follow-up assessment, which was available for a minority of patients (see online supplementary table S2).
C9P group patients with ALS had a younger age of onset (p=0.044), death (p=0.003) and shorter onset to death interval (p=0.035) relative to C9N ALS cases. Additionally, Kaplan–Meier log rank analysis found a significantly shorter survival in C9P ALS (figure 2; log-rank λ2=4.183, p=0.041). Site of onset data was available for 26 C9P group patients with ALS, and this group consisted of similar numbers of bulbar (7/26), cervical (8/26) and lumbar (10/26) onset cases, with 1 thoracic-onset case and no significant difference in onset site relative to C9N ALS.
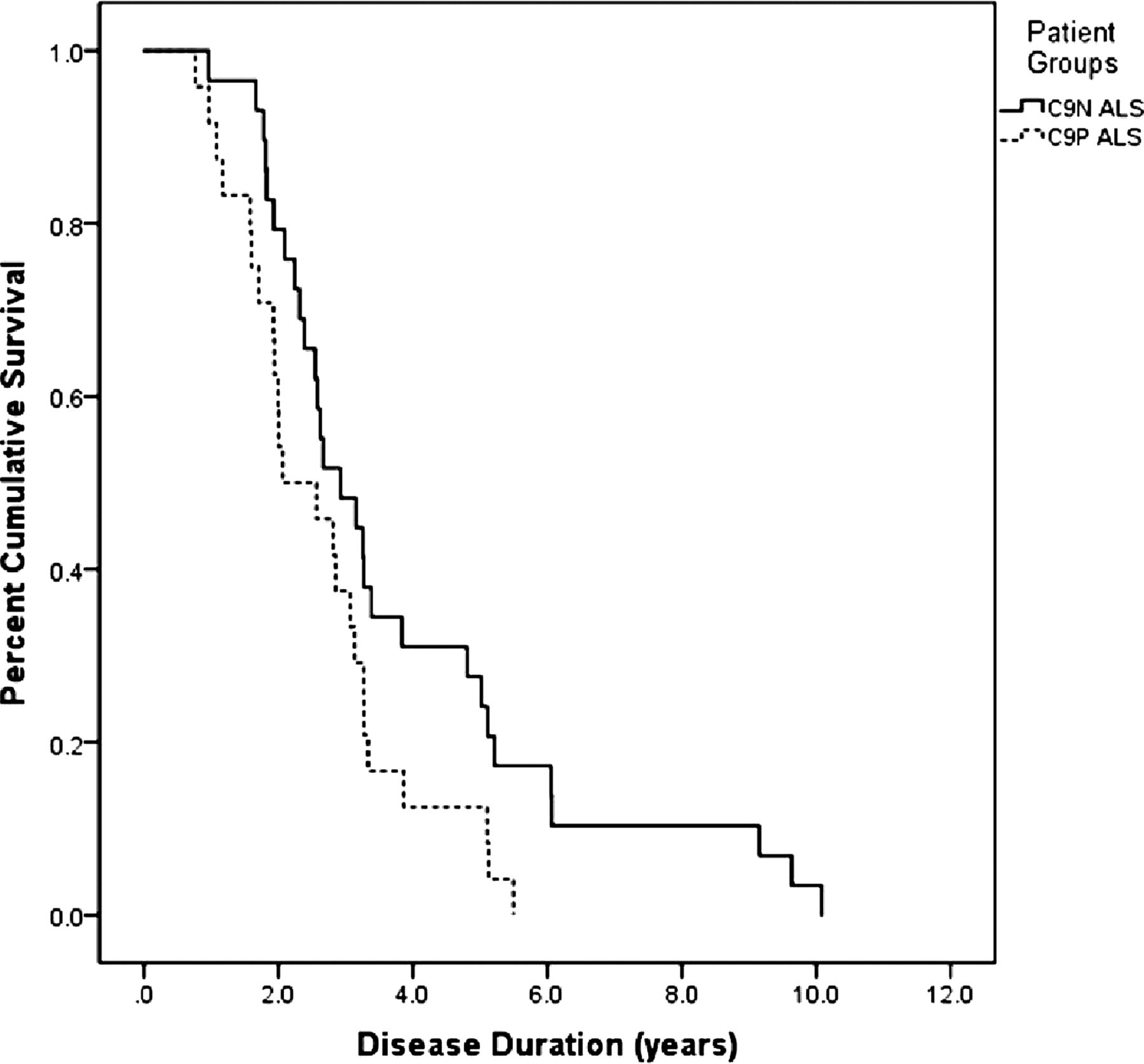
Kaplan–Meier curve analysis of survival of patients with amyotrophic lateral sclerosis with (C9P=dashed line) and without (C9N=solid line) the pathogenic C9orf72 hexanucleotide expansion (log-rank λ2=4.183, p=0.041).
A total of 36 C9P group patients with ALS and 24 C9N group patients with ALS had neuropsychological data available. Mean baseline MMSE (29.3 C9P, 29.4 C9N) and verbal fluency (11.9 C9P, 12.0 C9N) were similar between C9P and C9N ALS groups. Longitudinal data was available for MMSE (C9P=5, C9N=6) and verbal fluency (C9P=9, C9N=11). Although C9P ALS cases had a greater mean annualised decline in letter fluency (1.8 words/year) compared to C9N ALS (−0.1 words/year), no statistical differences in annualised decline were found in these small groups of ALS cases (see online supplementary table S2).
Neuroimaging analysis
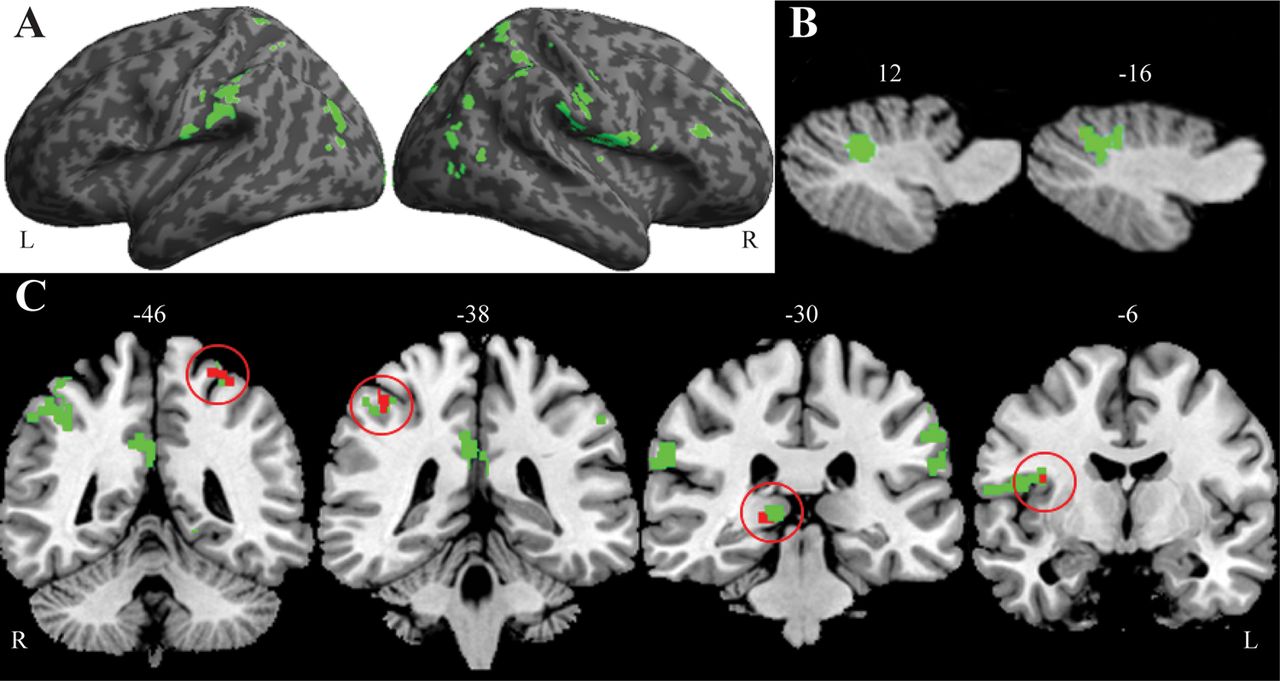
MRI data were available for 41 FTLD cases (14 C9P, 27 C9N). There were no significant demographic differences between patient subgroups and older-aged controls (see online supplementary table S3). Permutation analysis of GM thickness found significant bilateral GM thinning for C9P and C9N relative to older-aged controls (see online supplementary table S4). Direct comparison of GM thickness revealed significantly greater thinning in C9P relative to C9N cases in right frontal, frontoinsular, cingulate, occipital and thalamic regions and bilateral parietal cortex (figure 3A, see also online supplementary table S4). The reverse comparison revealed no significant GM thinning for patients in the C9N relative to the C9P group. An analysis of the cerebellum found two significant clusters of reduced GM density in C9P compared to C9N (figure 3B, see also online supplementary table S4) with no significant clusters in the reverse analysis.
{kind=link}
{kind=link}
{kind=link}
Green regions illustrate areas of significant cortical atrophy in C9P compared directly to C9N frontotemporal lobar degeneration (FTLD) for cerebrum (A) and cerebellum (B). Coronal slices (C) illustrate bilateral parietal and right frontal and thalamic regions (green) that have significant atrophy in C9P FTLD, and a subset (red) that correlate with poor verbal fluency performance. Numbers indicate sagittal (B) and coronal (C) slice axes location.
Regression analysis revealed a significant relationship between poor baseline verbal fluency performance and GM thinning in right frontoinsular, inferior-parietal and thalamic regions (figure 3C, see also online supplementary table S4). We observed no relationship between verbal fluency and GM density in the cerebellum.
Neuropathological examination
A total of 25 patients with FTLD from our cohort were followed to autopsy (C9P=13, C9N=12). Cases did not differ by post-mortem interval or brain weight (see online supplementary table S5). Similar to neuroimaging findings of GM thinning in C9P FTLD, semiquantitative analysis found more severe neuronal loss and gliosis in mid-frontal cortex (p=0.022, p=0.005, respectively) and trends for thalamus and angular gyrus in C9P FTLD compared with C9N FTLD (table 3). As we described previously,25 there was no appreciable granule cell loss or gliosis observed in the cerebellum. C9P and C9N FTLD did not differ significantly in TDP-43 severity or cortical distribution. However, we found a predominance of TDP harmonised,33 subtype B (10/13; A=2/13, C=1/13), and hippocampal sclerosis was present in only one case (see online supplementary table S5). UBQLN inclusion burden was more severe in C9P FTLD for all cortical regions sampled and also in the granule layer of the cerebellum where there was almost exclusive involvement in C9P cases.
Pathological analysis of neurodegeneration in frontotemporal lobar degeneration (FTLD) with and without the C9orf72 repeat expansion
Comparison of C9P (n=12) and C9N (n=27) ALS cases showed equally low levels of extramotor cortical neuronal loss, gliosis and TDP-43 inclusions (data not shown), and a detailed report of UBQLN burden in ALS has been previously described.25
Due to our imaging finding of increased frontal, parietal, cerebellar and thalamic atrophy in C9P FTLD cases we performed additional tests to see if these were related to pathological markers of neurodegeneration. UBQLN severity in the mid-frontal cortex and angular gyrus correlated with neuronal loss and gliosis in these regions (all r>0.5, p<0.05; angular gyrus/neuronal loss r=0.399, p=0.054) and inversely correlated with overall post-mortem brain weight (all r<−4.8, p<0.05). TDP-43 pathology in these regions did not correlate with neuronal loss in the mid-frontal cortex, angular gyrus, or post-mortem brain weight (p>0.05).
We also evaluated whether reduced verbal fluency in C9P FTLD was related to pathological markers of neurodegeneration in these regions. Baseline verbal fluency score inversely correlated with TDP-43 severity in the mid-frontal cortex (r=−0.731, p=0.016) and UBQLN in the angular gyrus (r=−0.706, p=0.022). Neither TDP-43 nor UBQLN severity in the thalamus or cerebellum correlated with verbal fluency (p>0.05).
Discussion
A comprehensive, multimodal evaluation of one of the largest reported C9P FTLD and ALS cohorts reveals that, relative to C9N, C9P FTLD cases had more rapid cognitive decline in verbal fluency and the MMSE and C9P ALS cases had a shorter survival. In C9P FTLD, direct correlations of performance with imaging and autopsy studies appear to relate this most closely to disease in frontal and parietal brain regions. By selecting C9N cases highly likely to have underlying FTLD-TDP neuropathology, we can speculate that these results specifically reflect the intrinsic effects of the pathogenic C9orf72 expansion.
The demographic characteristics of our C9P group patients are similar to previous reports with a predominance of bvFTD clinical phenotype,9–11 ,19 ,27 ,28 a small number of clinically diagnosed AD cases,11 ,19 ,34 ,35 and many cases with significant family history.6–10 12–14 ,19 ,25 ,27 ,28 ,35 ,36
Previous cross-sectional analyses found marked executive dysfunction,27 ,28 in C9P FTLD. While our data showed poor baseline cognitive performance for C9P FTLD and closely matched cases of C9N FTLD, quantitative longitudinal neuropsychological testing provides novel evidence to suggest an increased rate of cognitive decline on a measure of executive functioning and a measure of global cognitive functioning in the majority of C9P FTLD cases. Further prospective detailed neuropsychological analysis of patients with C9P will help confirm these findings.
Our neuroimaging analyses indicate a likely neuroanatomical substrate for these clinical observations in C9P. Previous studies describe similar patterns of cortical11 ,19 ,28 ,37 and subcortical19 ,37 atrophy in C9P FTLD, including prominent parietal lobe and cerebellar involvement, and some reports note hemispheric symmetry compared with other hereditary forms of FTLD.19 ,28 ,37 Our findings also show bilateral disease compared to controls (see online supplementary table S4). However, we found more prominent right-sided disease in the direct comparison of C9P to C9N FTLD. Furthermore, there were no areas of increased atrophy in C9N relative to C9P FTLD, suggesting more severe disease burden in C9P FTLD. These selective differences in C9P may reflect in part our careful selection of C9N cases, as we minimised confounds associated with unrelated neuropathologies (ie, FTLD-tau and AD) that can be found in clinically defined FTD (ie, C9N) reference cohorts,15 ,20 used in a previous comparative study.37 Additionally, other studies limited comparisons largely to healthy controls,19 ,28 or qualitative assessments alone.11 ,27 Significant posterior parietal disease is an atypical neuroimaging feature in sporadic and other hereditary forms of FTLD,37 suggesting a unique pathophysiology of C9P FTLD that could serve as a potentially useful biomarker for the C9orf72 expansion. Although verbal executive measures such as letter-guided naming fluency are mediated bilaterally,38 ,39 our regression analysis found associations with mainly right sided areas, including the right frontoinsular region. This may reflect in part the asymmetric burden of disease in our direct comparison of C9P and C9N FTLD.
The analysis of neuronal loss and gliosis in the autopsy cases compliments our neuroimaging findings, as these areas of GM atrophy were also evident in the frontal, parietal and thalamic regions of patients studied at autopsy. In addition, we found UBQLN inclusions to be more severe in C9P cases and this correlated well with measures of neurodegeneration. Furthermore, TDP-43 inclusions in the frontal lobe and UBQLN severity in parietal regions correlated with poor verbal fluency performance. While several years intervened between clinical evaluation and death, the observation of similar findings in imaging studies obtained at the same time as cognitive measures supports the clinicopathological association we found. Together, these correlations appear to implicate frontal and parietal regions in declining cognition observed in C9P FTLD. It is intriguing that UBQLN pathology was more closely correlated to neuronal loss than TDP-43. This may indicate that UBLN pathology is a further downstream event in the neurodegenerative process in C9P and may contribute to the areas of increased cortical thinning seen in the voxel-based morphometry analysis of C9P FTLD compared with C9N FTLD. Future detailed clinicopathological correlations and cellular and animal-model experiments are required to clarify these interactions in the complex pathophysiology of C9P FTLD and ALS.
Patients with slowly progressive or late onset C9P FTD,8 ,11 ,19 ,28 ,40 and cases with minimal cortical atrophy,10 ,11 ,28 have been described. Indeed, individual analysis of our C9P FTLD cohort reveals a wide range of age at onset (44–69) and survival (1.5–12.7 years). However, the majority of individual patients tested had substantial annualised decline in MMSE and letter fluency (see online supplementary table S1), and all imaged patients had significant cortical atrophy on qualitative inspection. Thus, our results suggest that most C9P FTLD appear to clinically progress more rapidly than other forms of FTLD. Further understanding of the pathogenicity of expansion length and possible anticipation effect is necessary to fully resolve observations of phenotypic heterogeneity.
The observed shorter disease duration and survival in C9P ALS has been reported by some groups,13 ,14 ,25 while others did not find this difference.36 The distribution of the site of onset in the C9P ALS group in our study was similar to the general ALS population, as in some previous reports,9 ,13 ,14 ,28 ,34 and contrary to others that report a majority of bulbar-onset phenotype.12 ,25 ,27 ,36 These discrepancies may be due in part to sample size and control patient selection. Indeed, we found marginal statistical significance in our analysis and these results will require confirmation in large prospective series. Some have also described more severe executive dysfunction in C9P ALS cases,14 corresponding to extramotor frontal cortical atrophy on MRI,14 and high rates of comorbid dementia.12 ,13 ,36 Our cohort of C9P ALS only showed a trend for greater annualised decline in letter fluency compared to C9N ALS. This may reflect inclusion of ALS-FTLD in the FTLD subgroup analyses, the small number of ALS cases with cognitive testing, or the potential relatively rapid rate of motor progression in ALS. Further study with larger, prospective ALS datasets would be beneficial to clarify these observations.
Our study is limited due to the retrospective nature of data collection and small sample sizes in some subgroup analyses. While we attempted to limit the C9N cases to TDP-43 proteinopathies, there may be patients who are non-deceased with other, co-occurring neuropathologies, as TDP-specific biomarkers are currently lacking. Keeping in mind these limitations and others mentioned above, our findings suggest that C9P may confer more rapid cognitive decline in the majority of FTLD and reduced survival in ALS. Our observations have significant implications for clinical trial design, as patients with C9P could influence outcome measures of cognition or survival.14 Future prospective longitudinal study of C9P will be crucial to confirm our findings here and help further characterise the clinical heterogeneity of cases. The potential impact of the C9orf72 hexanucleotide repeat on prognosis, and its occurrence in apparently sporadic cases, will be of utmost importance to elucidate for clinical practice and for the evaluation and implementation of emerging disease-modifying treatments.
Acknowledgments
Correction notice This paper has been amended since it was published Online First. In table 1, in the row “Mean (SEM) age of onset”, in the column for p values, a typographical error was inserted. In the ALS row, p value 0.444 should be 0.044.
Acknowledgments
Acknowledgements We would like to thank the patients and their families for their kind contributions to make this study possible.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online tables
Footnotes
-
Contributors DJI: data analysis and interpretation and drafting and critical revision of the manuscript. CTM, JB, JP, DJL, JB, KR, JBT, AB, KC, EMW, LS, JHW, PAC, DAW, VMV, LFM, LE: acquisition and analysis of data and interpretation, administrative support and critical revision of the manuscript. SEA, VML, JQT and MG: obtained funding for support and provided critical revision of the manuscript and MG provided the concept and design and overall supervision of the study.
-
Funding This study was supported by the NIH P30AG010124, P01 AG017586, R01 NS44266, R01 AG15116, P01 AG32953, P01 NS53488, R21 NS06311 and the Wyncote Foundation. DJI is supported by the NIH T32-AG000255, CTM is support by HD060406, JB is supported by a grant of the Deutsche Forschungsgemeinschaft DFG (AOBJ586910) and JBT is supported by a grant of the Alfonso Martín Escudero Foundation.
-
Competing interests DAW has received research support from NIH (R01 AG037376, R01 MH086492, K23 AG028018, P30 AG010124), GE Healthcare and Pfizer, Inc. He has served as a consultant for GE Healthcare and Leclair Ryan. He received speaking honoraria from the American Academy of Neurology. SAE reports board membership for Cowan Group, Eli Lilly and BMS; consulting services for Philadelphia district attorney's office and Bonner Kiernan Trebach & Cociata LLP; grants from American College of Radiology Imaging Network, Pfizer, Alzheimer's Disease Neuroimaging Initiative, Johnson & Johnson, National Institute of Drug Abuse, Neuronetrix, National Institute of Mental Health, Eli Lilly; payments for lectures at Harvard University, Rush University Medical Center, Trinitas Regional Medical Center and University of Puerto Rico. VML and JQT report single consulting services to Pfizer, J&J, MetLife and BMS; royalty payments through Penn licenses; and research support from AstraZeneca and BMS. DJI, CTM, JB, DJL, AB, JP, JB, KC, EMW, JBT, LS, KR, JHW, PAC, VMV, LFM, LE and MG have no potential financial relationships of activities within the previous 3 years that could appear to have influenced the current work.
-
Ethics approval Ethics approval was provided by the Perelman School of Medicine at the University of Pennsylvania.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Clinical data used in this study is available in the CNDR integrated neurodegenerative disease database at the Perelman School of Medicine at the University of Pennsylvania. (Please see Xie SX, Baek Y, Grossman M, et al. Building an integrated neurodegenerative disease database at an academic health center. Alzheimers Dement 2011 7(4):e84–93.).