Article Text

Abstract
Objective In multifocal motor neuropathy (MMN), the destruction of the blood-nerve barrier (BNB) has been considered to be the key step in the disease process. The purpose of the present study was to ascertain whether sera from patients with MMN can open the BNB, and which component of patient sera is the most important for this disruption.
Methods We evaluated the effects of sera from patients with MMN, patients with amyotrophic lateral sclerosis, and control subjects on the expression of tight junction proteins and vascular cell adhesion molecule-1 (VCAM-1), and on the transendothelial electrical resistance (TEER) in human peripheral nerve microvascular endothelial cells (PnMECs).
Results The sera from patients with MMN decreased the claudin-5 protein expression and the TEER in PnMECs. However, this effect was reversed after application of an anti-vascular endothelial growth factor (anti-VEGF) neutralising antibody. The VEGF secreted by PnMECs was significantly increased after exposure to the sera from patients with MMN. The sera from patients with MMN also increased the VCAM-1 protein expression by upregulating the nuclear factor kappa-B (NF-κB) signalling. The immunoglobulin G purified from MMN sera decreased the expression of claudin-5 and increased the VCAM-1 expression in PnMECs.
Conclusions The sera from MMN patients may disrupt the BNB function via the autocrine secretion of VEGF in PnMECs, or the exposure to autoantibodies against PnMECs that are contained in the MMN sera. Autoantibodies against PnMECs in MMN sera may activate the BNB by upregulating the VCAM-1 expression, thereby allowing for the entry of a large number of circulating inflammatory cells into the peripheral nervous system.
- NEUROPATHY
- BLOOD-BRAIN BARRIER
- IMMUNOLOGY
- NEUROCHEMISTRY
- NEUROMUSCULAR
Statistics from Altmetric.com
Introduction
Multifocal motor neuropathy (MMN) is an acquired neuropathy characterised by chronic or stepwise progressive asymmetrical limb weakness without sensory defects.1 ,2 The etiopathogenesis of MMN is not well known, but there is some evidence that the disease has an immunological basis, because immunological therapies including high-dose intravenous immunoglobulins (IVIg) show therapeutic effects, although corticosteroids and plasma exchange are largely ineffective.3–8 Anti-GM1 IgM antibodies can be found in some patients with MMN,2 ,9 but it is unclear whether these antibodies are pathogenic. However, some reports support the hypothesis that autoantibodies that bind to gangliosides activate the classical complement system pathway and induce nerve injury by the incorporation of the complement membrane attack complex (C5b-9) in peripheral motor nerves.10–12
The blood-nerve barrier (BNB) protects the nerve fibres in the PNS from systemic inflammatory reactions and immune responses.13 ,14 Several lines of evidence have demonstrated that the disruption of the BNB, causing the leakage of macromolecules like immunoglobulin and cytokines, is a key step in the disease process of chronic inflammatory demyelinating polyneuropathy (CIDP).15 A few reports about the pathological findings in the motor nerves of a patient with MMN suggested that the disruption of the BNB may occur during the disease process of MMN.16–18 However, it has not been adequately explained whether the sera from patients with MMN can disrupt the BNB, and which component of the patients’ sera is the most critical for the dysregulation of the BNB.
The purpose of the current study was to demonstrate the effects of sera from patients with MMN on the impairment of the BNB function, and to clarify the roles of humoral factors, especially antibodies against the human BNB-composing endothelial cells, in the destruction of the BNB.
Materials and methods
Sera
This study was approved by the review boards of Tokushima University and Yamaguchi University following the principles of the Declaration of Helsinki. All patients consented to participate in this study. The acute-phase sera were collected from 11 patients with MMN who were diagnosed at Tokushima University Hospital or Yamaguchi University Hospital (table 1). All 11 patients met the clinical criteria for possible MMN based on the 2010 EFNS/PNS guideline19 and had an objective clinical improvement following IVIg treatment. Three of the 11 patients with MMN (patient nos. 4, 6, 7) were positive for anti-GM1 IgM antibodies (table 1). The sera from nine patients with definite amyotrophic lateral sclerosis (ALS) diagnosed by the El Escorial criteria20 were also used in this study as disease controls. The sera from 10 healthy individuals served as normal controls. Blood samples were taken before treatment and stored at −80°C until use. All sera were incubated at 56°C for 30 min just prior to use.
Clinical profiles and nerve conduction data of patients with MMN
Cell culture and treatment
The immortalised human peripheral nerve microvascular endothelial cells (PnMECs), which were named ‘FH-BNB’, were generated previously.14 ,21 The PnMECs were treated with culture medium containing 10% patient or healthy control sera in a humidified atmosphere of 5% CO2/air. PnMECs treated with culture medium with 10% fetal bovine serum (FBS; Sigma, St. Louis, Missouri, USA) were used as controls. The transendothelial electrical resistance (TEER) value was measured 24 h later, and the total proteins were obtained the next day.
Reagents
The culture medium for PnMECs was previously described.21 Polyclonal anti-claudin-5 and anti-occludin antibodies were purchased from Zymed (San Francisco, California, USA). The polyclonal anti-actin and anti-nuclear factor kappa-B (anti-NF-κB) p65 antibodies were obtained from Santa Cruz (Santa Cruz, California, USA). The polyclonal anti-IL-1β, anti-TNF-α, anti-TGF-β, anti-vascular endothelial growth factor (anti-VEGF), anti-IL-6, and anti-vascular cell adhesion molecule-1 (VCAM-1) antibodies were purchased from R&D Systems (Minneapolis, Minnesota, USA). The broad-spectrum matrix metalloproteinase (MMP) inhibitor, GM6001, was purchased from Chemicom (Temecula, California, USA).
Western blot analysis
The protein samples (10–20 μg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; Bio-Rad), and then were transferred to nitrocellulose membranes (Amersham, Chalfont, UK) as described previously.21 The membranes were treated with relevant antibodies (dilution 1 : 100) for 2 h as the primary antibodies and then incubated with secondary antibodies (dilution 1 : 2000) for 1 h at room temperature. The membranes were visualised by enhanced chemiluminescence detection (ECL-prime, Amersham, UK). A densitometric analysis was performed using the Quantity One software program (Bio-Rad, Hercules, California, USA).
TEER studies
The TEER values of cell layers were measured with a Millicell electrical resistance apparatus (Endohm-6 and EVOM, World Precision Instruments, Sarasota, Florida, USA) as described previously.21 The PnMECs were seeded (1×106 cells/insert) on the upper compartment and incubated with each type of medium (non-conditioned medium used as a control, conditioned medium contained 10% patient sera) for 24 h.
Permeability studies
The PnMECs were grown to confluence on 24-well tissue culture inserts (0.4 μm pore size, 1.0×104 cells/insert) as described previously.14 About 1 300 μL of the culture medium was added to the lower well, and 500 μL of culture medium containing sodium fluorescein (10 μg/mL) of molecular weight 400 kDa was added to the upper compartment of each insert. After incubation for 15, 30, 45 or 60 min at 37°C, the lower chamber was sampled and the fluorescence was measured using an MX3000P instrument (Stratagene).
Quantitative analysis of VEGF by ELISA
The serum levels of VEGF were determined in triplicate by an ELISA using commercially available kits (R&D Systems, Minneapolis, Minnesota, USA). The results were expressed as picograms of VEGF per millilitre (pg/mL), based on the standards provided with the available kits.
Treatment with neutralising antibodies
The sera from patients with MMN were pretreated with either a neutralising antibody (2.0 μg/mL) against IL-1β, TNF-α, TGF-β, IL-6, or VEGF or normal rabbit IgG (control Ab) for 6 h at 4°C. PnMECs were cultured with the sera from three patients with MMN containing each neutralising antibody at 37°C.
Treatment with an MMP inhibitor or NF-κB inhibitor
A broad-spectrum MMP inhibitor, GM6001 (Chemicom, Temecula, California, USA), or NF-κB activation inhibitor (Calbiochem, Darmstadt, Germany) was prepared for the inhibition study. The sera from patients with MMN were pretreated with 25 μM of GM6001 or 150 nM of the NF-κB inhibitor for 12 h at 37°C. PnMECs were cultured with the sera from each of three patients with MMN with GM6001 or the NF-κB inhibitor.
IgG purification from serum
The IgG fractions were obtained from the sera of five patients with anti-GM1 antibody-negative MMN or five healthy individuals by affinity chromatography using a Melon Gel IgG Spin Purification Kit (Thermo Scientific, Rockford, Illinois, USA). Cells were treated with culture medium containing either purified patient or healthy individual IgG (final concentration 400 μg/mL). Cells treated with culture medium containing purified IgG obtained from FBS (Sigma, final concentration 400 μg/mL) were used as controls.
Data analysis
An unpaired, two-tailed Student t test was used to determine the significance of differences between the means of two groups. A p value <0.01 was considered to be statistically significant.
Results
MMN sera decreased the BNB function
Table 1 shows the clinical profiles and nerve conduction data for each of the patients with MMN. We first examined whether the sera from patients with MMN affects the BNB function. The amount of claudin-5 in the PnMECs was significantly decreased after exposure to sera from patients with MMN, whereas it was not affected by the sera from patients with ALS or healthy controls, as determined by a Western blot analysis (figure 1A–D). The amount of occludin protein was not changed after exposure to sera from patients MMN or ALS, or healthy controls (figure 1E). The TEER value of PnMECs was significantly decreased, and the sodium fluorescein (NaF) permeability of PnMECs was significantly increased, after exposure to sera from patients with MMN, although it was not changed by incubation with sera from patients with ALS or healthy controls (figure 1F,G). The presence of anti-GM1 IgM antibodies did not influence either the change of claudin-5 protein amounts or the NaF permeability of the PnMECs (figure 1H,I).


(A–C) The effects of the sera of patient with multifocal motor neuropathy (MMN) on the tight junction proteins in human peripheral nerve microvascular endothelial cells (PnMECs) as determined by a Western blot analysis. The changes of claudin-5 and occludin in PnMECs were determined after exposure to the sera from patients with MMN or amyotrophic lateral sclerosis (ALS), or from healthy controls. (D and E) Each bar graph reflects the combined densitometry data from each independent experiment. The amount of claudin-5 protein in PnMECs was significantly decreased after exposure to the sera from patients with MMN (mean±SEM, n=11, p<0.01). The amounts of claudin-5 and occludin were not significantly affected by exposure to the sera from patients with ALS (mean±SEM, n=9) or from healthy controls (mean±SEM, n=10). (F and G) The transendothelial electrical resistance value of PnMECs was significantly decreased (F) and the NaF permeability of PnMECs was significantly increased (G) after exposure to MMN sera, but these were not influenced by exposure to sera from patient with ALS or healthy controls. (H and I) The effect of anti-GM1 IgM antibodies in the sera from patients with MMN on the amount of tight junction proteins and NaF permeability. The amount of claudin-5 protein was decreased, and the NaF permeability was increased after exposure to the sera from patients with MMN with and without anti-GM1 IgM antibodies, compared to that of control, irrespective of the presence of anti-GM1 antibody. Therefore, the presence of anti-GM1 IgM antibodies did not influence the claudin-5 protein amounts (H) or the NaF permeability (I). Control: non-conditioned DMEM containing 20% fetal bovine serum (FBS); MMN: conditioned medium with 10% serum from a patient with MMN diluted with non-conditioned DMEM containing 10% FBS; ALS: conditioned medium with a 10% concentration of serum from a patient with ALS diluted with non-conditioned DMEM containing 10% FBS; Normal: conditioned medium with 10% serum from a healthy control diluted with non-conditioned medium of DMEM containing 10% FBS; GM1-IgM positive MMN, conditioned medium with 10% serum samples of patients with MMN with anti-GM1 IgM antibodies; GM1-IgM negative MMN, conditioned medium with 10% serum samples of patients with MMN without anti-GM1 IgM antibodies.
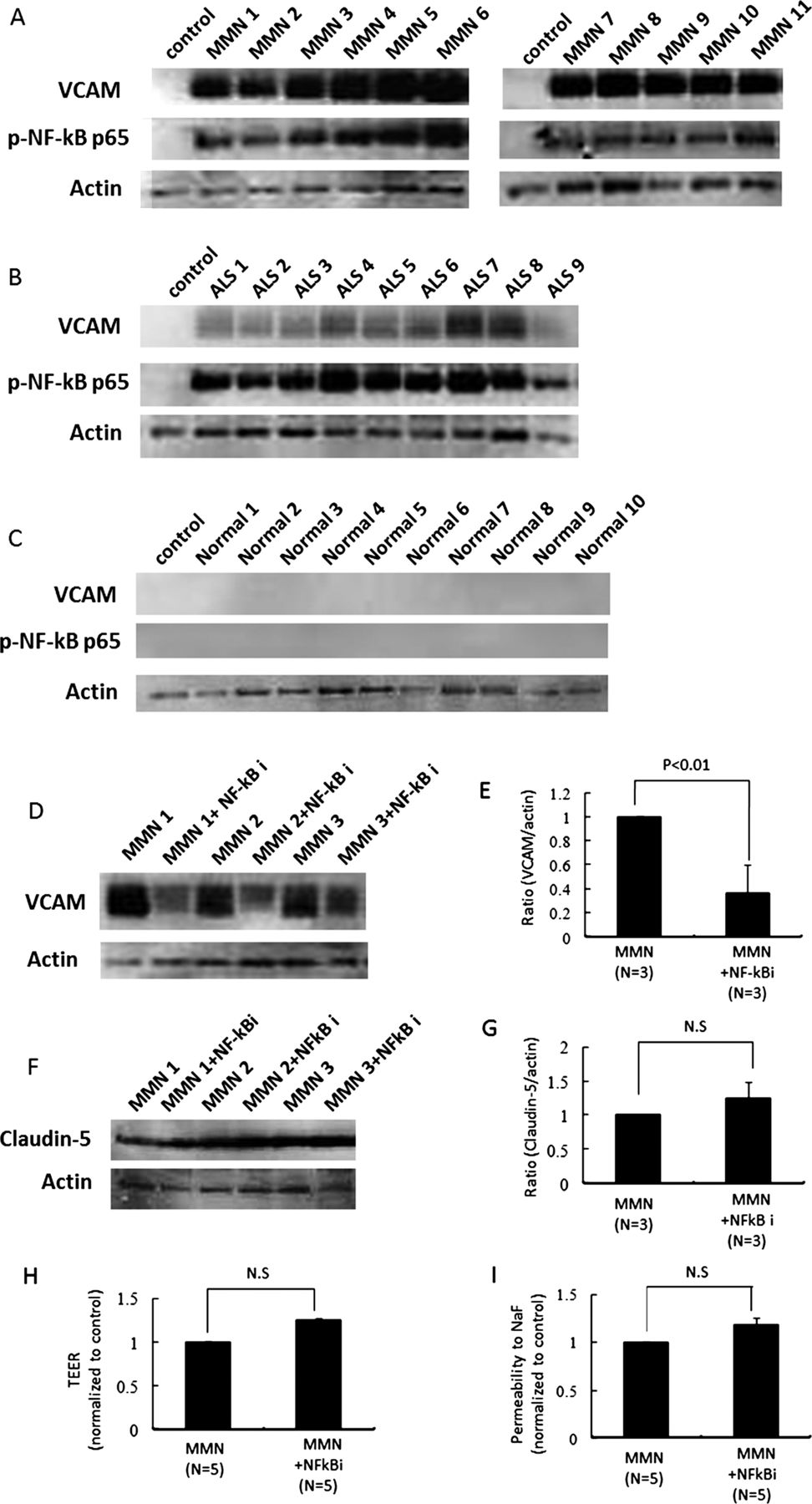
MMN sera increased the amount of VCAM-1 protein through NF-κB signalling in PnMECs
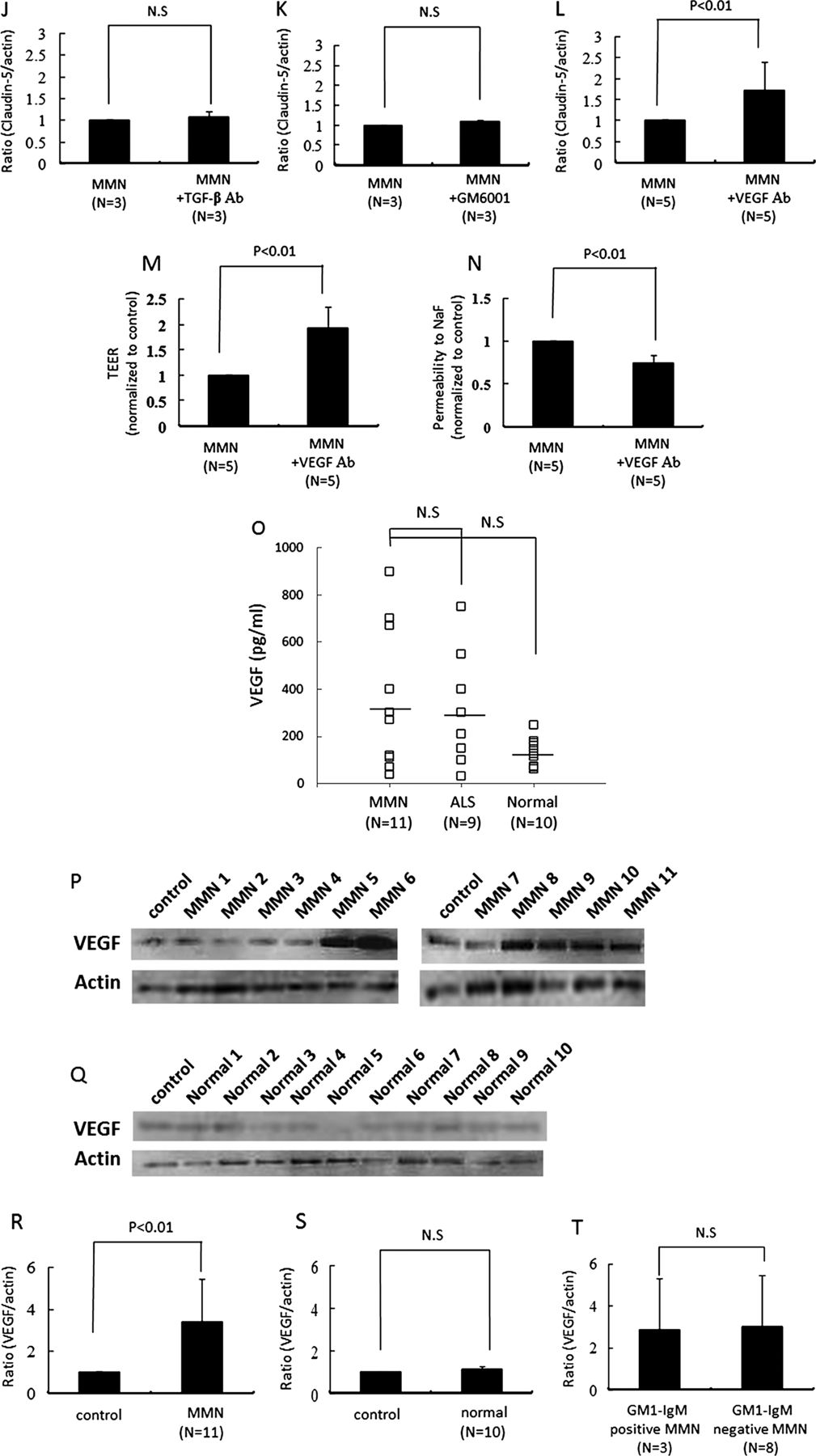
We next analysed whether the sera from patients with MMN affect the expression of adhesion molecule. The amount of vascular cell adhesion molecule-1 (VCAM-1) and NF-κB p65 protein in PnMECs was significantly increased after exposure to sera from patients with MMN or ALS, whereas it was not changed by the sera from healthy controls, as determined by a Western blot analysis (figure 2A–C). To clarify the contribution of NF-κB to the BNB breakdown, we investigated the amount of VCAM-1 and claudin-5 protein, the TEER value and the NaF permeability in PnMECs after MMN sera exposure with or without pretreatment with NF-κB inhibitor. The amount of VCAM-1 protein in PnMECs after exposure to MMN sera after pretreatment with the NF-κB inhibitor was significantly decreased compared with that in cells without pretreatment with the NF-κB inhibitor (figure 2D,E). The amount of claudin-5 protein, the TEER value and the NaF permeability in PnMECs after MMN sera exposure and pretreatment with the NF-κB inhibitor were not changed compared with those in cells without treatment with the NF-κB inhibitor (figure 2F–I).

(A–C) The effects of sera on the amount of adhesion molecules in human peripheral nerve microvascular endothelial cells (PnMECs) were determined by a Western blot analysis. The changes in the amount of VCAM-1 protein in PnMECs were determined after exposure to the sera from patients with multifocal motor neuropathy (MMN) (A) or amyotrophic lateral sclerosis (ALS) (B), or from healthy controls (C). (D) The effects of an NF-κB inhibitor on the expression of adhesion molecules in PnMECs after exposure to the sera from a patient with MMN was determined by a Western blot analysis. The amount of VCAM-1 protein in PnMECs after MMN sera exposure in cells pretreated with the NF-κB inhibitor was significantly decreased compared to that of cells without NF-κB inhibitor pretreatment. (E) Each bar graph reflects the combined densitometry data from independent experiments (mean±SEM, n=3, *: p<0.01). (F–I) The effects of the NF-κB inhibitor on the amount of claudin-5 protein, the transendothelial electrical resistance (TEER) value and the NaF permeability of PnMECs after exposure to the sera from a patient with MMN. (F) The amount of claudin-5 protein in PnMECs after MMN sera exposure and pretreatment with the NF-κB inhibitor was not changed compared to that of cells without NF-κB inhibitor pretreatment. (G) Each bar graph reflects the combined densitometry data from independent experiments (mean±SEM, n=3, p<0.01). (H and I) The TEER value and NaF permeability across PnMECs after MMN sera exposure in cells pretreated with the NF-κB inhibitor were not significantly changed compared to those of cells without NF-κB inhibitor pretreatment. Control: non-conditioned DMEM containing 20% fetal bovine serum (FBS); MMN: conditioned medium with 10% serum from a patient with MMN diluted with non-conditioned DMEM containing 10% FBS; ALS: conditioned medium with a 10% concentration of serum from a patient with ALS diluted with non-conditioned DMEM containing 10% FBS; Normal: conditioned medium with 10% serum from a healthy control diluted with non-conditioned medium of DMEM containing 10% FBS; MMN+NF-κB inhibitor: conditioned medium with 10% MMN sera pretreated with the NF-κB inhibitor.
MMN sera disrupted the BNB through the upregulation of autocrine VEGF in PnMECs
To clarify the contribution of inflammatory cytokines or MMPs to the BNB breakdown in MMN, the TNF-α, IL-1β, IL-6, TGF-β or VEGF activities were neutralised using the corresponding neutralising antibodies or MMPs were inhibited by the broad-spectrum MMP inhibitor, GM6001 (figure 3A–F). The amount of claudin-5 protein in PnMECs was significantly increased after exposure to the MMN sera pretreated with the anti-VEGF neutralising antibody, as determined by a Western blot analysis (figure 3F,L), whereas it did not change after preincubation with TNF-α, IL-1β, IL-6 or TGF-β neutralising antibodies or GM6001 (figure 3A–E,G–K). The TEER value of the PnMECs was also significantly increased, and the NaF permeability of PnMECs was significantly decreased after exposure to MMN sera pretreated with the anti-VEGF antibody (figure 3M,N). The serum concentration of VEGF did not significantly differ between the patients with MMN , patients with ALS and healthy controls as groups, although some patients with MMN and ALS had VEGF concentrations higher than the range observed in the healthy controls, as determined using an ELISA method (figure 3O). We thus considered that MMN sera may disrupt the BNB by increasing the autocrine secretion of VEGF in PnMECs. The expression of VEGF in PnMECs was found to be significantly increased after exposure to sera from patients with MMN (figure 3P,R), whereas it did not change after exposure to the sera from healthy controls (figure 3Q,S). The presence of anti-GM1 IgM antibodies did not influence the changes in the amounts of VEGF proteins in the PnMECs (figure 3T).
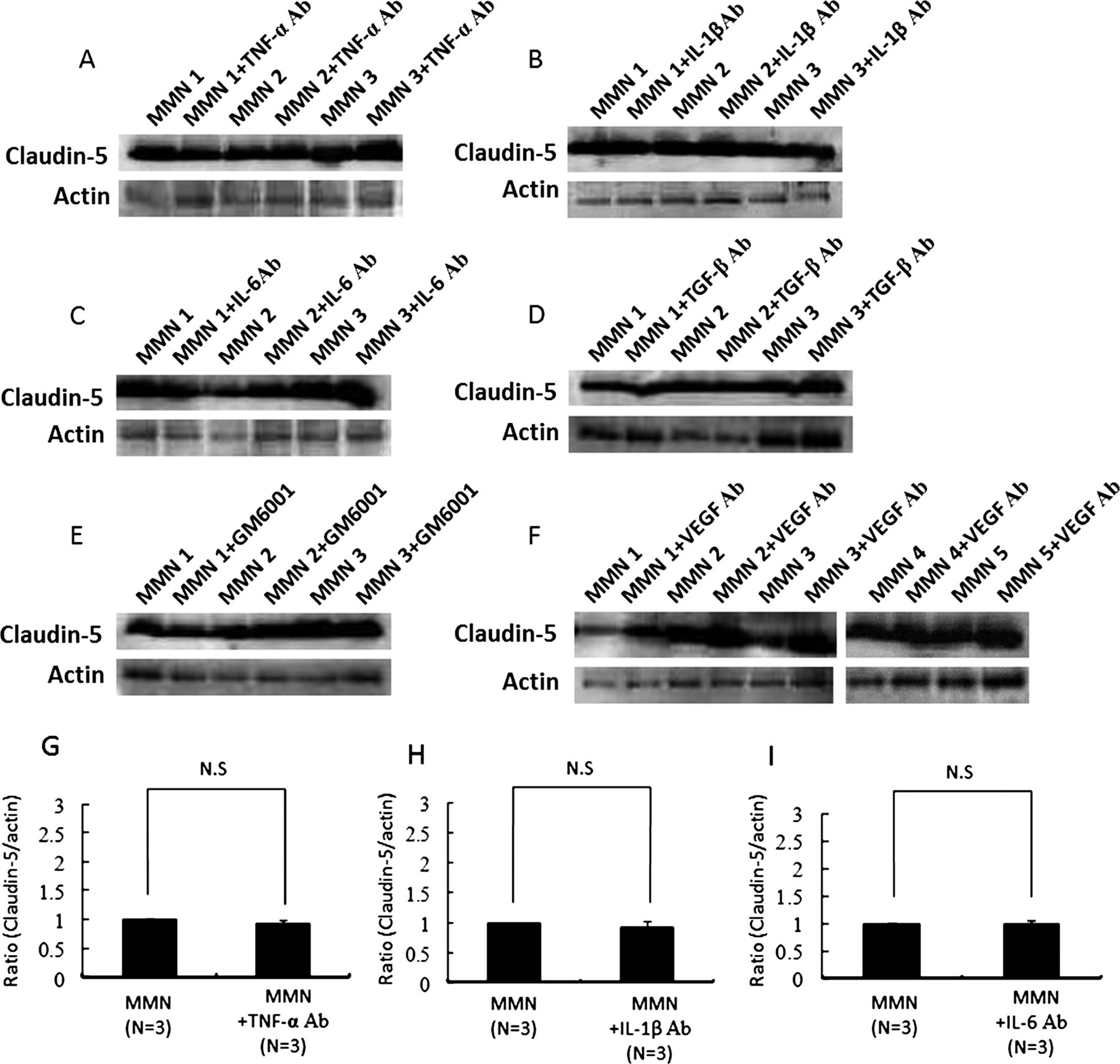

(A–F) The effects of anti-TNF-α, IL-1β, IL-6, TGF-β or vascular endothelial growth factor (VEGF) neutralising antibodies or a matrix metalloproteinase inhibitor on the amount of tight junction proteins in human peripheral nerve microvascular endothelial cells (PnMECs) after exposure to the sera from patients with multifocal motor neuropathy (MMN) was determined by a Western blot analysis. (G–L) Each bar graph reflects the combined densitometry data from each independent experiment (mean±SEM, n=3, *: p<0.01). (F and L) Preincubation with an anti-VEGF neutralising antibody increased the amount of claudin-5 protein in PnMECs (mean±SEM, n=5, *: p<0.01). The transendothelial electrical resistance value of PnMECs significantly increased (M) or the NaF permeability of PnMECs significantly decreased (N) after incubation with the sera from patients with MMN that were pretreated with an anti-VEGF neutralising antibody (mean±SEM, n=5). (O) The serum VEGF concentration was analysed in patients with MMN or amyotrophic lateral sclerosis, or from healthy control subjects. The bars indicate the mean of each group. No significant differences were observed between the three groups. (P–T) The expression of VEGF by PnMECs after exposure to the sera from patients with MMN. The amount of VEGF protein in the PnMECs was significantly increased after exposure to the sera from patients with MMN (P), although it did not change after exposure to the sera from healthy controls (Q). (R and S) Each bar graph reflects the combined densitometry data from each independent experiment (mean±SEM, MMN n=11, healthy control n=10, p<0.01). (T) The presence of anti-GM1 IgM antibodies did not influence the changes in the amounts of VEGF proteins in the PnMECs. MMN: conditioned medium with 10% MMN sera diluted with DMEM containing 10% fetal bovine serum (FBS); MMN+TNF-α Ab: conditioned medium with 10% MMN sera pretreated with an anti-TNF-α neutralising antibody; MMN+IL-1β Ab: conditioned medium with 10% MMN sera pretreated with an anti-IL-1β neutralising antibody; MMN+IL-6 Ab: conditioned medium with 10% MMN sera pretreated with an anti-IL-6 neutralising antibody; MMN+TGF-β Ab: conditioned medium with 10% MMN sera pretreated with an anti-TGF-β neutralising antibody; MMN+GM6001: conditioned medium with 10% MMN sera pretreated with a GM6001; MMN+VEGF Ab: conditioned medium with 10% MMN sera pretreated with an anti-VEGF neutralising antibody. Control: non-conditioned DMEM containing 20% FBS; MMN: conditioned medium with 10% serum from a patient with MMN diluted with non-conditioned DMEM containing 10% FBS; Normal: conditioned medium with 10% serum from a healthy control diluted with non-conditioned medium of DMEM containing 10% FBS; GM1-IgM positive MMN: conditioned medium with 10% serum samples of MMN patients with anti-GM1 IgM antibodies; GM1-IgM negative MMN: conditioned medium with 10% serum samples of MMN patients without anti-GM1 IgM antibodies.
Purified serum IgG from patients with MMN disrupts the BNB
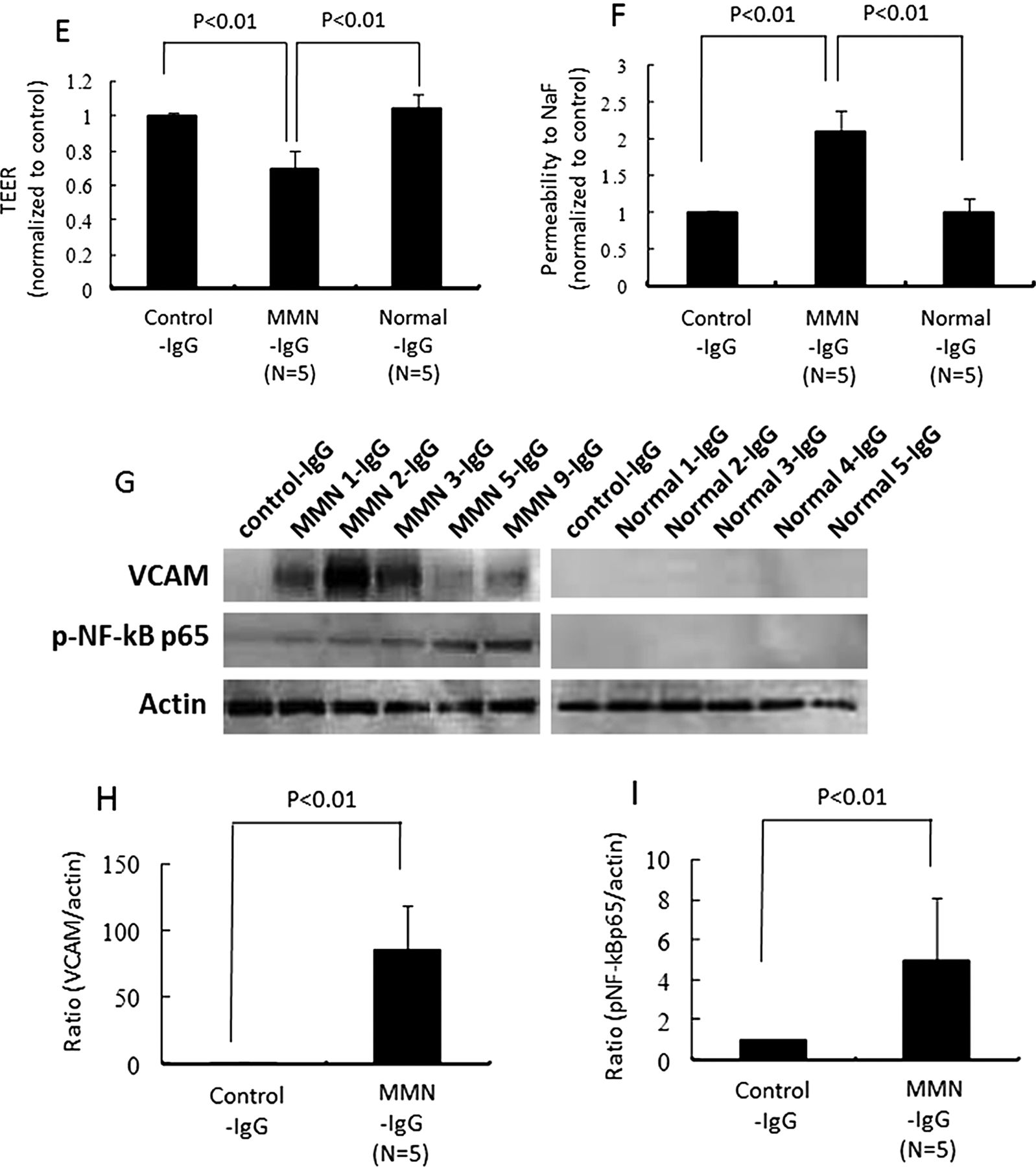
We next analysed whether autoantibodies against human PnMECs were present in the purified IgG fractions of sera from patients with MMN by a Western blot analysis. Antibodies that bound to PnEMCs were detected in the purified IgG fractions of sera from five patients with MMN patient, which predominantly reacted with one or more antigens of approximately 30, 45, 50, 54, 56 and 70 kDa in PnMEC lysates (figure 4A). Notably, antibodies against the antigens corresponding to 54, 56 and 70 kDa were specific for patients with MMN and were not seen in the sera from patients with ALS or healthy controls (figure 4A). The lower molecular bands corresponding to 30, 45 and 50 kDa in PnMECs were not specific for patients with MMN, because these bands were commonly detected in ALS patients or healthy controls (figure 4A). We next examined whether the purified serum IgG from patients with MMN, rather than anti-GM1 IgM antibodies, was indeed responsible for the disruption of the BNB. The amount of claudin-5 in PnMECs was significantly decreased after exposure to the purified IgG fractions of sera from anti-GM1 IgM antibody-negative MMN patients, whereas it was not affected by the purified IgG fractions from healthy controls, as determined by a Western blot analysis (figure 4B,C). The amount of VEGF proteins did not change following exposure to the purified serum IgG fractions obtained from the patients with MMN and healthy controls (figure 4B,D). The TEER value of PnMECs was significantly decreased and the NaF permeability of PnMECs was significantly increased after exposure to the purified IgG fraction from anti-GM1 IgM antibody-negative MMN patients, although it was not changed by incubation with the purified IgG fractions from healthy controls (figure 4E,F). In addition, the amount of VCAM-1 and NF-κB p65 in PnMECs was significantly increased after exposure to the purified IgG fraction from anti-GM1 IgM antibody-negative MMN patients, whereas it was not changed by the purified IgG fractions from healthy controls, as determined by a Western blot analysis (figure 4G–I).
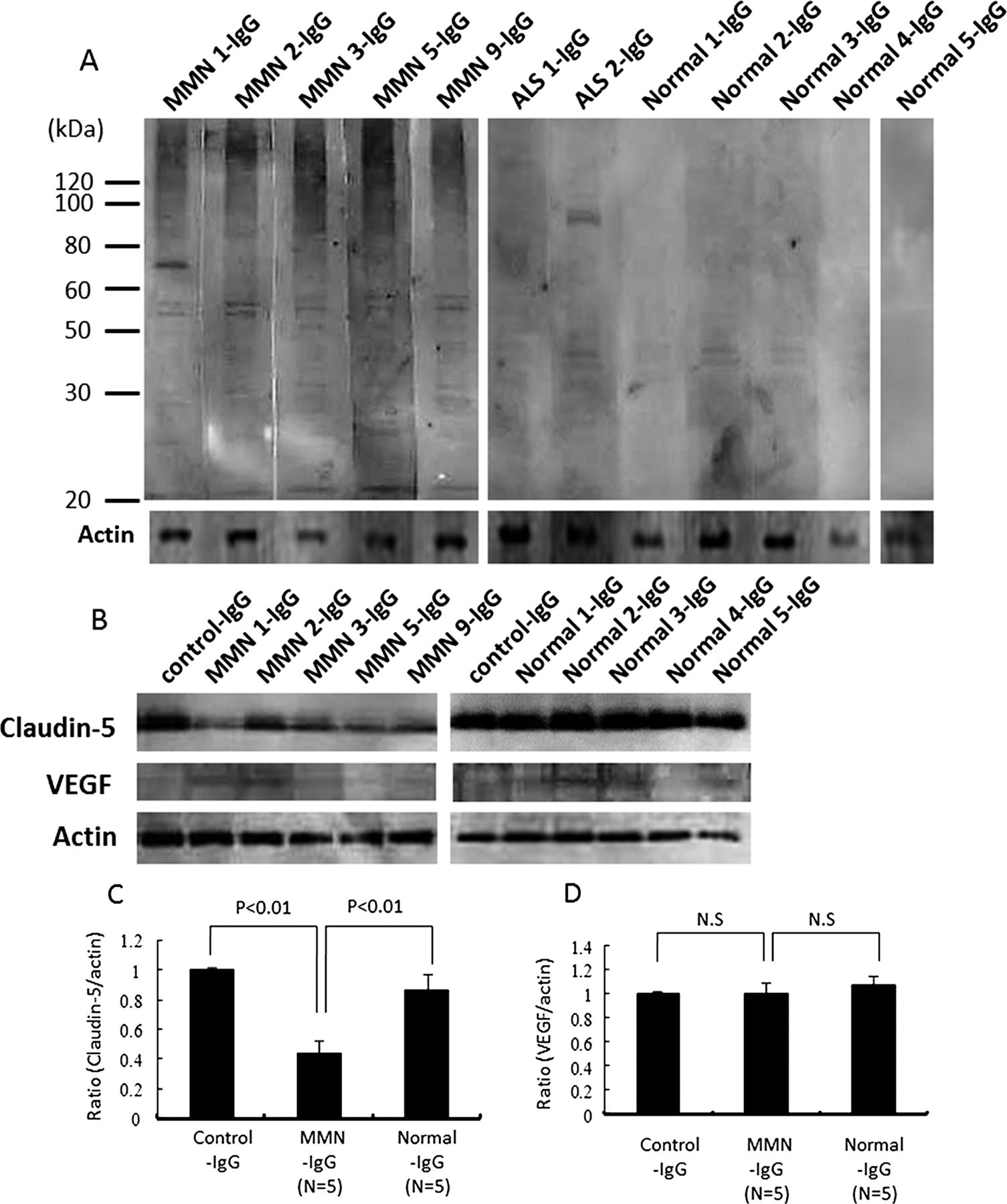

(A) Representative results obtained by immunoblotting of human peripheral nerve microvascular endothelial cell (PnMEC) lysates. The blots were exposed to the purified serum IgG from five patients with multifocal motor neuropathy (MMN), two patients with amyotrophic lateral sclerosis (ALS) and five healthy controls after loading 20 mg of protein lysates from PnMECs. The purified IgG fractions of the sera from patients with MMN predominantly reacted with one or more antigens of approximately 30, 45, 50, 54, 56 and 70 kDa in the PnMEC lysates. The purified serum IgG samples from the two patients with ALS also reacted with approximately 30, 40, 45, 50 and 90 kDa antigens of PnMECs. The two bands corresponding to the 40 and 45 kDa antigens of PnMECs were detected from the purified IgG fractions from the five healthy controls. The expression of actin was used as an internal standard. (B and C) The effects of the purified serum IgG from patients with MMN without anti-GM1 IgM antibodies on the expression of tight junction proteins and vascular endothelial growth factor (VEGF) in human peripheral nerve microvascular endothelial cells (PnMECs) were determined by a Western blot analysis. (B) The amount of claudin-5 in PnMECs was significantly decreased after exposure to the purified IgG fractions of patient's sera, whereas it was not affected by the purified IgG fractions from healthy controls, as determined by a Western blot analysis. The amount of VEGF proteins did not change following exposure to the purified serum IgG fractions obtained from the patients with MMN and healthy controls. (C and D) Each bar graph reflects the combined densitometry data from independent experiments (mean±SEM, n=5, *: p<0.01). The transendothelial electrical resistance value of PnMECs was significantly decreased (E) and the NaF permeability of PnMECs was significantly increased (F) after exposure to the purified IgG fraction from patients with MMN, although it was not changed by incubation with the purified IgG fractions from healthy controls. (G) The amount of VCAM-1 and NF-κB p65 in PnMECs was significantly increased after exposure to the purified IgG fraction from patients with MMN, whereas it was not changed by the purified IgG fractions from healthy controls, as determined by a Western blot analysis. (H and I) Each bar graph reflects the combined densitometry data from independent experiments (mean±SEM, n=5, p<0.01). Control-IgG: conditioned medium containing purified IgG fractions obtained from fetal bovine serum; MMN-IgG: conditioned medium with containing purified IgG fractions obtained from the sera of patients with MMN; ALS-IgG: conditioned medium with containing purified IgG fractions obtained from the sera of patients with ALS; Normal-IgG: conditioned medium with containing purified IgG fractions obtained from the sera of healthy individuals.
Discussion
The etiopathogenesis of MMN has not been clarified. Some evidence suggests that the disease has an immunological basis, primarily due to the occurrence of clinical improvement following the administration of immunological therapy, including high-dose IVIg.3–7 However, the precise mechanisms and target antigens of this immune response are unknown. An important diagnostic feature is the presence of persisting multifocal partial conduction blocks (CBs) that selectively affected the motor axons in the nerve conduction studies.3–8 Although the pathological basis of CBs is considered to be focal demyelination, this has rarely been confirmed in MMN by morphological studies, because tissue samples taken from the motor nerves of patients with MMN are extremely rare. Some previous reports on sensory nerve biopsies in patients with MMN have described either normal findings or unspecific changes, consistent with the infrequent sensory impairment in patients with MMN.22 ,23 Only a few reports on motor nerve biopsies or autopsies in MMN cases have been published. For example, Kaji et al16 described the myelinated axons and the formation of onion bulbs with endoneurial oedema and perineurial thickening in the medial pectoral nerve biopsy at the site of CB and suggested that impaired remyelination caused by the disruption of the BNB was the mechanism responsible in this case. Oh et al17 reported perivascular lymphocytic infiltration in the endoneurial or perineurial microvessels of the BNB in the motor nerves from an autopsy case of a patient with MMN. Thus, a leaky BNB that allows the intrusion of circulating pathogenic antibodies and inflammatory cytokines may play a crucial role in the development of MMN. Some studies have indeed demonstrated anti-GM1 antibody-mediated focal demyelination and blockade of voltage-dependent Na+ channels at the node of Ranvier in vivo and in vitro24–26 and reported that the sera obtained from patients with MMN can block nerve conduction in distal motor nerves in mice.27 However, the molecular mechanism of BNB breakdown in MMN has not been adequately explained as yet. In the present study, we used conditionally immortalised human BNB-derived endothelial cells to analyse the effects of the sera from patients with MMN on the impairment of the BNB function.14 We have also previously reported that VEGF disrupts the BNB and that sera obtained from patients with Bickerstaff's brainstem encephalitis and Miller Fisher syndrome did not influence the barrier function in the same in vitro BNB model.21 ,28 Our present study is the first to demonstrate that the sera from patients with MMN can disrupt the BNB. The expression of claudin-5 and the TEER values were decreased, and the NaF permeability of PnMECs was increased after exposure to the MMN sera. Together, these results indicate that humoral factors in the MMN sera disrupt the BNB. We therefore first tried to identify the most important substance involved in disrupting the BNB in patients with MMN.
The presence of circulating cytokines, including TNF-α, IL-1β and VEGF, appears to be linked to the pathogenesis of the BNB breakdown in patients with MMN. Recent data suggest that these cytokines can disrupt the BNB; in particular, VEGF was able to induce BNB impairment.21 Our present study demonstrated that the BNB function was restored after adding a neutralising anti-VEGF antibody to the MMN sera, indicating that VEGF was the key molecule responsible for the disruption of the BNB in the patients with MMN in our study. Although the serum concentration of VEGF was not increased in the patients with MMN compared to that from healthy controls, the secretion of VEGF by PnMECs was increased after exposure to the MMN sera. This finding suggests that the effect of VEGF occurred via an autocrine mechanism; thus, minimal secretion may lead to a significant effect. Our present studies demonstrated that the neutralising anti-VEGF antibody may also have therapeutic potential for restoring the BNB integrity in MMN. We were unable to identify which humoral factors in the MMN sera caused the increased VEGF secretion observed in the present study; however, we demonstrated that the amount of VEGF proteins did not change following exposure to IgG obtained from the MMN sera in our study, thus indicating that unknown humoral factors other than IgG in the MMN sera are key mediators of increased VEGF secretion (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
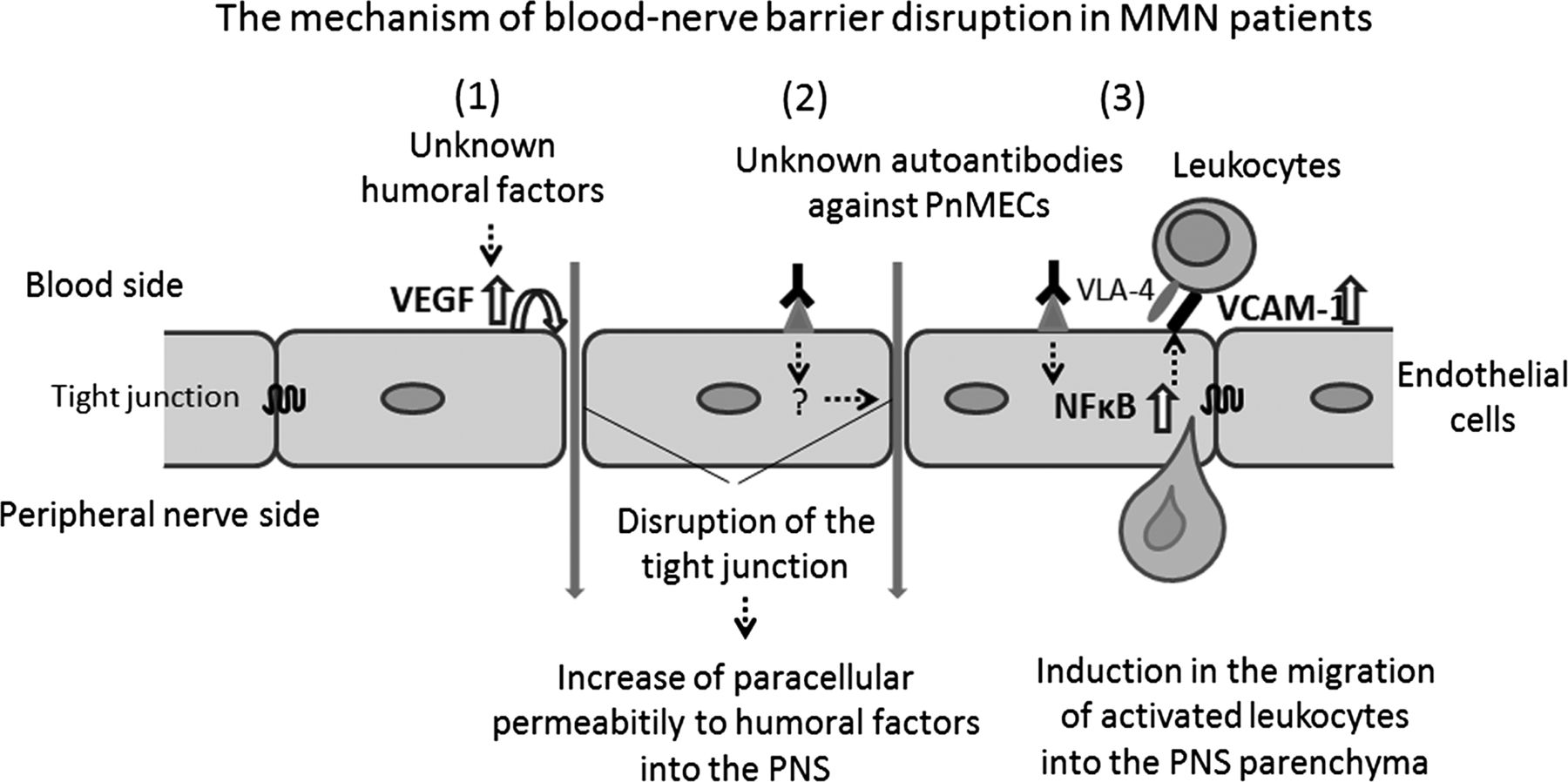
Schematic depiction of the assumed molecular mechanisms underlying the blood-nerve barrier (BNB) disruption observed in patients with multifocal motor neuropathy (MMN). The disruption of the BNB caused by humoral factors present in the sera of patients with MMN involves two differently regulated steps, including the disruption of tight junction proteins via the autocrine secretion of vascular endothelial growth factor from PnMECs induced by unknown humoral factors (1) and exposure to unknown autoantibodies against PnMECs (2), and the upregulation of VCAM-1 via NF-κB signalling in PnMECs induced by exposure to unknown autoantibodies against PnMECs in MMN sera (3).
We next hypothesised that antibodies binding to PnMECs might be involved in the BNB disruption in patients with MMN, because antibody-mediated immunological therapies including high-dose IVIg are effective against MMN. We thus determined whether purified IgG from the MMN sera without complement would have a direct influence on the BNB properties. Our results demonstrated that the purified IgG from the MMN sera decreased the amount of claudin-5 and the TEER value, and increased the permeability of the BNB, thus indicating that unknown antibodies, possibly IgG, against PnMECs from the MMN sera cause the disruption of the BNB (figure 5). This finding supports our hypothesis concerning the etiopathogenesis of MMN: MMN has an antibody-mediated immunological basis.
The proportion of patients with MMN with anti-GM1 antibodies in our present study (∼27%), was lower than that reported in several previous studies, in which these antibodies were detected in 22–85% of patients with MMN.2 The wide variation observed in the incidence of these antibodies is likely derived from the different ELISA assays used in the different studies.2–6 We believe that this fact did not influence the possible consequences of the interpretation of our present study because the absence of these antibodies does not exclude a diagnosis of MMN. The frequent presence of anti-GM1 IgM antibodies in the sera of patients with MMN and their decrease during improvement induced by cyclophosphamide29–31 have favoured the hypothesis that GM1, which is present on the endothelial cells forming the BNB, may be the target of this immune response. GM1 is indeed present on the endothelial cells forming the BNB,32 and anti-GM1 monoclonal antibodies can open the bovine BNB without the help of complement in vitro.33 However, we found that the presence of anti-GM1 IgM antibodies in the patients’ sera did not influence the BNB function in the PnMECs. Although the anti-GM1 IgM antibody is still a candidate cause of the disruption of the BNB, we consider that untested factors or unknown antibodies against PnMECs in the sera of patients with MMN other than the anti-GM1 IgM antibodies may be the key players that upset the BNB.
The interaction of VCAM-1 and very late activating antigen-4 (VLA-4) has a unique role in the pathogenesis of multiple sclerosis because it is involved in rolling and the arrest of leucocytes, which is a prerequisite for the activation of all further steps of transendothelial leucocyte migration.34 ,35 We demonstrated that the sera from patients with MMN increased the amount of VCAM-1 protein, and this effect was reversed by exposure to an NF-κB inhibitor. The present study also has shown that the sera from patients with ALS had increased amounts of VCAM-1 protein. This finding can be explained by the hypothesis concerning a complicated pathogenesis of ALS, wherein immunological factors, including cytokines, chemokines and MMPs, and the disruption of the blood-brain barrier (BBB) and the blood-spinal cord barrier may play key roles in the development of the disease.36 ,37 We also have indicated that the purified IgG of the sera from patients with MMN increased the amount of VCAM-1 proteins. This indicated that the unknown antibodies against PnMECs in the sera from patients with MMN may increase the VCAM-1 protein expression by upregulating the NF-κB signalling, thus causing the migration of activated leucocytes to the PNS parenchyma (figure 5), supporting the previous observation that perivascular lymphocytic infiltration in endoneurial microvessels of the BNB was present in autopsy cases of MMN.17 ,18 Natalizumab is a humanised monoclonal antibody against the VLA-4 and inhibits the binding of leucocytes to the VCAM-1 expressed on activated brain vessels.38 ,39 Our results provide the theoretical basis for applying natalizumab clinically in patients with MMN. In case of CIDP, natalizumab cannot be recommended at present, because Wolf et al40 reported the case of a patient with CIDP in whom natalizumab treatment was not beneficial. Novel therapy directed specifically towards the reduction of VCAM-1 in the BNB could also be a possible therapeutic strategy for the treatment of MMN.
In conclusion, our study demonstrated that the disruption of the BNB caused by the humoral factors present in the sera of patients with MMN involves two differently regulated steps; the disruption of BNB function via the autocrine secretion of VEGF from PnMECs induced by unknown humoral factors or exposure to unknown autoantibodies against PnMECs, and the up-regulation of VCAM-1 in PnMECs induced by exposure to unknown autoantibodies against PnMECs in MMN sera. These data may provide novel explanations concerning the etiopathogenesis and the triggers of the BNB breakdown in MMN. A further analysis of the molecular mechanisms underlying the BNB breakdown observed in MMN, including the identification of the unknown molecules responsible for the disruption of BNB, and clarification of the effects of natalizumab and neutralising anti-VEGF antibodies against the disruption of the BNB, using an ex vivo or in vivo experimental model would assist in the development of therapies for this disabling disease.
References
Footnotes
-
Contributors FS performed the experiments, analysed and interpreted the data and wrote the manuscript. YS, TM and AT performed experiments and analysed the data. MO, NM and AM recruited patients. MK and RK evaluated the data and edited the manuscript. TK conducted and supervised the study, evaluated the data and wrote the manuscript.
-
Funding This work was supported in part by research grants (Nos. 24790886, 22790821 and 23659457) from the Japan Society for the Promotion of Science, Tokyo, Japan, by research grant (K2002528) from Health and Labor Sciences Research Grants for research on intractable diseases (Neuroimmunological Disease Research Committee) from the Ministry of Health, Labor and Welfare of Japan and also by the Translational Research Promotion Grant from Yamaguchi University Hospital.
-
Competing interests All authors declare no potential conflicts of interest.
-
Patient consent Obtained.
-
Ethics approval The study was approved by the ethics committee of Yamaguchi University.
-
Provenance and peer review Not commissioned; externally peer reviewed.