Article Text

Abstract
Objective To classify the patterns of functional decline in patients with sporadic amyotrophic lateral sclerosis (ALS) and explore the genetic backgrounds that modified these patterns.
Methods We included 465 patients with sporadic ALS in the analysis and clustered the longitudinal functional scores in the registered patients, using a mixture approach of a non-linear mixed-effects model. We conducted a genome-wide analysis of 572 983 single nucleotide polymorphisms (SNPs). We then assessed the association between the clusters of longitudinal functional scores and SNPs.
Results We identified the following four clusters of longitudinal functional decline in the cases: a rapid decline cluster, an intermediate decline cluster, a sigmoidal decline cluster and a moderate decline cluster. We identified seven SNPs associated with the rapid decline cluster, using a recessive model (p=3.47–8.34×10−8). The OR for the probabilities of the rapid decline cluster ranged from 5.5 to 5.84. Homozygosity for the minor alleles in the seven SNPs, which constituted a linkage disequilibrium (LD) block, was associated with decreased expression of TTN (encoding Titin, a large sarcomere protein) in the expression quantitative trait loci database of a large-scale Japanese genetic variation database (p=8.6×10−10–1.1×10−7). TTN expression in immortalised lymphocyte lines was decreased in patients who were homozygous for the minor alleles compared with those who were homozygous for the major alleles (n=19 in each group, p=0.002).
Conclusions We detected an LD block associated with a rapid functional decline in patients with sporadic ALS, which is linked to decreased expression of TTN.
Statistics from Altmetric.com
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease characterised by progressive loss of upper and lower motor neurons.1 ,2 Approximately 5% of patients with ALS have a family history of the disease, and an increasing number of genes in which single mutations can cause ALS have been identified.3 Although the mutations that cause familial ALS also account for a subset of sporadic ALS cases, the majority of cases of sporadic ALS are thought to be multifactorial. To investigate the genes associated with sporadic ALS, a number of genome-wide association studies (GWAS) using large-scale genotyping of single nucleotide polymorphisms (SNPs) have been conducted.4 Unfortunately, the reported associations were not sufficiently strong to develop disease models and have been proven to be difficult to replicate.5
To date, a number of clinical trials for ALS therapies have failed to show effectiveness,6 ,7 and satisfying disease-modifying therapeutics do not exist for ALS. Previous studies have indicated that the course of functional decline in patients with ALS varies significantly between individuals,8–10 which may reduce the power of analysis in clinical trials for ALS. Several clinical factors, such as old age at onset or bulbar onset, have been shown to be related to a steeper functional decline; however, the genetic markers associated with the disease course of sporadic ALS have not been identified. The gene polymorphisms or mutations that are associated with the disease course can be the target for disease-modifying therapies and stratification in clinical trials.
To explore the genetic variants that modify the disease course of sporadic ALS, we classified the progression patterns of functional decline in patients with ALS and conducted a GWAS, using a multicentre prospective cohort of patients with ALS. We detected the SNPs associated with a rapid functional decline in patients with sporadic ALS, which were linked to decreased expression of TTN. Titin encoded by TTN is a large sarcomere protein and plays important roles in skeletal muscle function. This study suggests that decreased expression of TTN may influence the disease progression, possibly through decreased skeletal muscle function, and TTN can be a therapeutic target to ameliorate the functional decline in patients with sporadic ALS.
Methods
Patient registry and follow-up system
We developed a multicentre registration and follow-up system referred to as the ‘Japanese Consortium for Amyotrophic Lateral Sclerosis research (JaCALS)’,11 ,12 which consisted of 30 neurology facilities in Japan. The patients with ALS who were diagnosed in these facilities were consecutively registered after written informed consent was obtained. The ethics committees of all the participating institutions approved the study. At registration, a full clinical examination was conducted by neurologists. To standardise the procedure and the examination, the three organising doctors (HaW, NA and RN) visited each participating facility and ascertained the evaluation methods for the study.
Disease onset was defined as the time when the patient initially became aware of muscle weakness or impairment in swallowing, speech or respiration. We enrolled patients who fulfilled the revised El Escorial criteria. The included patients were prospectively followed up with telephone surveys conducted by clinical research coordinators (CRCs) or via examinations by neurologists, every 3 months, and the degree of deterioration in muscle function was determined at each time point. We used the Japanese version of the ALS functional rating scale (ALSFRS-R), which was validated by Ohashi et al as a scale of motor function. We developed a telephone survey system in which the CRCs conducted a telephone survey every 3 months that referenced the ALSFRS-R flow charts (Japanese version). We previously confirmed the reliability of this telephone survey system,13 using a method similar to that used to confirm the English version of the telephone survey in several studies.14 ,15 Prior to the study, we informed the CRCs of the study plan, the procedures for the telephone survey, ethical issues relevant to the study, and the requisite considerations for patients with ALS and caregivers. Moreover, we provided the participants with general information regarding ALS. The introduction of tracheostomy positive-pressure ventilation (TPPV) or the death of a patient was defined as the end point, and TPPV-free survival was defined as survival. The observation period continued until 5 years after the onset of the disease or until the end point.
Patients
A total of 652 patients with sporadic ALS were registered in JaCALS from January 2006 to December 2012. After we screened for known gene mutations, we excluded nine patients owing to the presence of the following gene mutations: superoxide dismutase-1 (SOD1) mutation (n=7); trans-active response DNA-binding protein 43 kDa (TDP-43) mutation (n=1); and TRK-fused gene (TFG) mutation (n=1). No C9orf72 mutations were identified in the included patients. A total of 643 patients were used to conduct the genome-wide genotyping. Seventy-three patients who were categorised as having a diagnosis of ‘suspected ALS’ according to the revised El Escorial criteria and 30 patients with TPPV at registration were excluded from the study. Subsequently, we excluded 34 patients for whom the disease duration had been over 5 years at registration and 41 patients with invalid clinical data. Overall, we included 465 patients with sporadic ALS in the study. Loss of patients during follow-up was addressed if the case was not followed up after July 2012, without attainment of an end point.
The clinical data of the registered patients were kept anonymous in each participating facility within the JaCALS study group and assigned unique patient numbers. The data were sent to the clinical data centre located at Nagoya University Graduate School of Medicine and entered into the JaCALS database.
The statistical methods for clustering the longitudinal ALSFRS-R scores using a mixture approach of the non-linear mixed-effects model are shown in the online supplementary methods.
Genome-wide genotyping and quality control
Genome-wide genotyping of the 643 patients registered in JaCALS was conducted using the HumanOmniExpressExome BeadChip (Illumina, San Diego, California, USA) at the Laboratory for Genotyping Development, Centre for Integrative Medical Sciences, RIKEN, in Yokohama, Japan. During quality control of the samples, seven samples with a genotype call rate of <98% were excluded from the analysis. Additionally, one sex-mismatched sample was identified and excluded from further analysis. To detect duplicate samples or relatives among the samples, we checked the samples in which the estimated genome-wide identity-by-descent (IBD) proportion of alleles shared was >0.1875. Principal component analysis (PCA) was conducted to identify the population outliers. We estimated IBD sharing using the PLINK option ‘--genome’ and the PCA using SMARTPCA on a linkage disequilibrium (LD)-pruned set of 103 363 SNPs, which was obtained by removing large-scale high-LD regions or SNPs with a genotype call rate <0.98, or a minor allele frequency (MAF) <0.01, or Hardy-Weinberg equilibrium (HWE) (p<1×10−6). LD pruning was performed using the PLINK option ‘--indep-pairwise 50 5 0.2’. Based on the IBD sharing, any family relatedness was identified and excluded from further analyses, including PCA. Based on the first 10 principal components, four population outliers were identified and excluded from further analyses. After quality control, a total of 13 samples were excluded. Among the 465 patients who were included in the clinical analyses of this study, 11 patients were excluded during quality control of the SNP typing. Therefore, 454 patients were included in the clinicogenotype association study.
Among the 951 117 SNPs, we excluded any SNPs with a genotype call rate<0.98, a MAF<0.01 or a HWE p<1×10−6. The remaining 572 983 autosomal SNPs were analysed in the GWAS.
Identifying SNPs associated with the disease progression phenotypes
We categorised the 465 patients into four groups based on the estimated membership values and subsequently compared the allele frequencies between the rapid decline cluster and the three other clusters, and between the moderate functional decline cluster and the three other clusters, to identify the SNPs associated with the functional decline phenotypes. For each SNP, we estimated the ORs, the 95% CIs and the p value for the dominant, recessive and additive models, using logistic regression analysis with Firth's correction. In this analysis, the significance level α was determined by dividing 0.05 by the number of analysed SNPs for a Bonferroni correction (α=0.05/572 983=8.73×10−8). Furthermore, a haplotype analysis to detect the LD for candidate SNPs with genome-wide significant p values was performed using Haploview 4.2 (http://www.broadinstitute.org/scientific-community/science/programs/medical-and-population-genetics/haploview/haploview).
Expression quantitative trait loci information
We obtained the expression quantitative trait loci (eQTL) information in the Japanese population from the Human Genetic Variation Database (HGVD) constructed by the 5 core facilities in Japan (HGVD Release V.1.41).16 The loci information is available at the following website: http://www.genome.med.kyoto-u.ac.jp/SnpDB/.
Lymphocytes isolated from the patients with ALS
Epstein-Barr virus (EBV)-immortalised lymphocyte lines were generated from B lymphocytes obtained from peripheral blood from all the patients with ALS at the time of registration. The lymphocytes obtained from 19 patients with age-matched and sex-matched ALS for each genotype were classified as minor homozygous, major homozygous or heterozygous, using the LD block with the candidate SNPs associated with the disease progression phenotype, and were applied to the gene expression analysis. The method of real-time quantitative PCR for gene expression analysis is shown in the online supplementary methods.
Results
Characteristics and longitudinal changes of total ALSFRS-R scores
The demographic and clinical characteristics of the patients are presented in table 1. The disease duration at registration ranged from 1.1 to 60.0 months. The average duration of follow-up was 2.5 years (SD 2.0), and 37 patients (8.0%) were lost to follow-up. The median TPPV-free survival time was 45.0 months (range 5.0 to 60.0 months). The Kaplan-Meier curve of TPPV-free survival is shown in online supplementary figure S1.
Clinical characteristics of the included patients (n=465)
Phenotypes of total ALSFRS-R score change
Figure 1A displays the trajectories of the longitudinal ALSFRS-R scores for each of the 465 patients. Using the ALSFRS-R of the 2476 time points for the 465 patients, we estimated the parameters using a Bayesian approach (see online supplementary table S1). Figure 1B shows the estimated mean curves of the four clusters for the longitudinal ALSFRS-R. The four clusters are: cluster 1—a rapid functional decline cluster (blue line); cluster 2—an intermediate functional decline cluster (red line); cluster 3—a sigmoidal functional decline cluster (yellow line) and cluster 4—a moderate functional decline cluster (green line). The identified trajectories were not linear but, rather, were curvilinear. Cluster 1 included patients whose score showed a rapid decline from disease onset to 24 months, followed by a steady period. Cluster 2 included patients whose score decreased obliquely. Cluster 3 included patients who showed a sigmoidal score decrease and cluster 4 included patients whose score decreased gradually or was maintained at a value close to that at the disease onset. We also calculated the membership values for each cluster and categorised the 465 patients using those values: 59 patients (13%) in cluster 1 (figure 1C), 113 patients (24%) in cluster 2 (figure 1D), 72 patients (15%) in cluster 3 (figure 1E) and 221 patients (48%) in cluster 4 (figure 1F).

Phenotypes of the total amyotrophic lateral sclerosis functional rating scale (ALSFRS-R) score change. (A) Trajectories of the ALSFRS-R scores of the 465 patients. (B) Mean curves for each cluster based on the non-linear mixed-effects model. (C–F) Longitudinal ALSFRS-R scores for each cluster. (C) Cluster 1, rapid functional decline cluster. (D) Cluster 2, intermediate functional decline cluster. (E) Cluster 3, sigmoidal functional decline cluster. (F) Cluster 4, moderate decline cluster.
Comparison of the clinical characteristics of the included patients with the phenotypes of the total ALSFRS-R score change showed that no clinical factors were significantly associated with the phenotypes (see online supplementary table S2).
Genetic analysis for associations between the disease progression phenotypes
We performed a genetic analysis to detect associations between the disease progression phenotypes; the rapid functional decline phenotype (cluster 1) and the moderate functional decline phenotype (cluster 4) of the ALSFRS-R were considered to have particular clinical importance. We compared the allele frequencies between the rapid functional decline cluster and the three other clusters, and between the moderate functional decline cluster and the three other clusters, to identify SNPs associated with the disease progression phenotypes. In the 454 patients who were included in the clinicogenotype association study, we compared the allele frequencies between the two groups, using dominant, recessive and additive models. The OR, 95% CI and p value were estimated. Using a threshold of 8.73×10−8 for the p value (based on the Bonferroni adjustments), we identified eight SNPs associated with the rapid functional decline cluster of ALSFRS-R in the recessive model (p=3.47–8.34×10−8, table 2 and figure 2A,B). Two of the eight SNPs were duplicated; therefore, seven SNPs were associated with the rapid decline ALSFRS-R cluster. The OR for the probabilities of the rapid functional decline cluster ranged from 5.50 to 5.84. The proportion of the rapid functional decline cluster in the patients with minor allele homozygosity of the SNPs ranged from 35.9% to 38.2%, in contrast to the range of 8.2–8.7% in the patients with major allele homozygosity and 10.1–10.7% in the patients with heterozygosity (table 2 and online supplementary table S3). An LD analysis revealed that the seven SNPs and the neighbouring area constituted an LD block (see online supplementary figure S2). The frequencies of homozygosity for the minor alleles and the major alleles in the all of the genotypes of the seven SNPs were 30% and 60%, respectively, which amounted to 90% of the observed haplotypes. In contrast, no SNPs were found to be associated with the moderate decline ALSFRS-R cluster.
SNPs significantly associated with a rapid decline of ALSFRS-R in the recessive model

Manhattan plot of the genome-wide association studies (GWAS). (A) Manhattan plot of the GWAS using the rapid functional decline cluster (cluster 1). The minimum p values among the additive, dominant and recessive models for each single nucleotide polymorphism (SNP) were plotted. The horizontal line represents the genome-wide significance level (α=8.73×10−8). (B) Regional association plot at 2q31.2. Minimum p values among the additive, dominant and recessive models for each SNP were plotted. The SNP with the minimum p value, rs4984020, is represented as a diamond. The other SNPs are coloured according to the extent of linkage disequilibrium within the SNPs. Genes within the regions are depicted by green arrows.
Partial imputation analysis
We performed an imputation analysis for the 7 Mb region surrounding rs4894020. The method of this analysis is shown in online supplementary methods, and the results are shown in online supplementary table S4 and figure S3.
TTN gene expression analysis
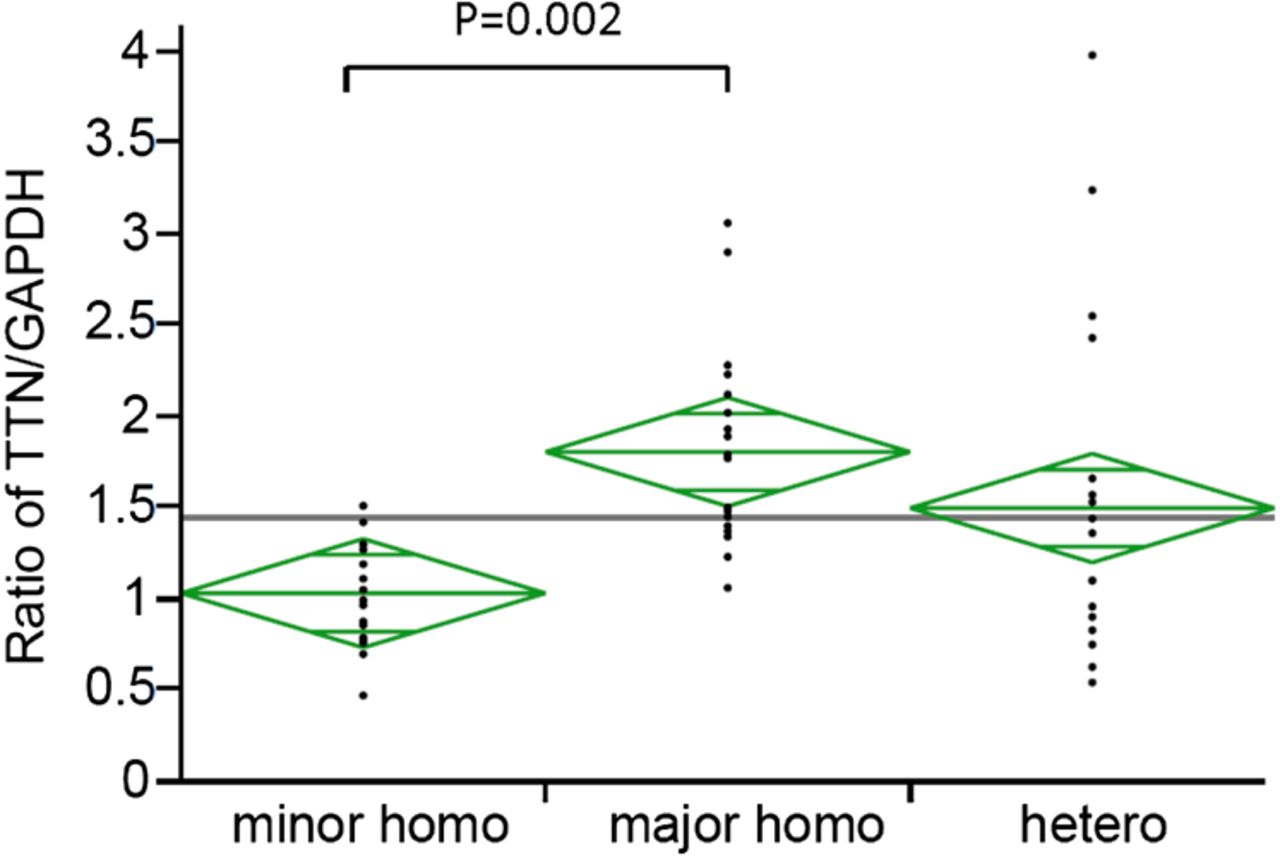
eQTL mapping of the HGVD demonstrated that all of the seven significant SNPs had an effect on TTN expression in lymphocytes (p=8.6×10−10–1.1×10−7, table 3). To confirm this effect in our patients with ALS, we measured the TTN expression levels in lymphocytes from patients with age-matched and sex-matched ALS with each genotype (minor homozygous, major homozygous and heterozygous) of the LD block, with the seven SNPs (see online supplementary table S5). The TTN messenger RNA (mRNA) expression level was measured via quantitative reverse transcription PCR and was found to be significantly lower in lymphocytes from patients who were homozygous for the minor allele (n=19) than in those who were homozygous for the major allele (n=19) (p=0.002, one-way analysis of variance and Tukey's test, figure 3).
eQTL data for the 7 SNPs associated with rapid decline on ALSFRS-R
{kind=link}
{kind=link}
{kind=link}
Relative expression of TTN in lymphocytes from patients with age-matched and sex-matched amyotrophic lateral sclerosis with each single nucleotide polymorphism genotype.
Discussion
This is the first report to classify the various courses of functional decline in patients with sporadic ALS and to identify the genetic factors associated with the rapid functional decline pattern.
As was observed in this study, consistent with a number of previous reports,8–10 the course of progression of the disease is highly variable among patients with sporadic ALS. Therefore, the factors that influence the differences in the clinical course are important for the development of a treatment for ALS. Various clinical factors concerning the progression and prognosis of patients with ALS have been reported. For example, old age at onset,17 onset with a bulbar symptom18 and poor nutritional status,19 have been shown to be associated with poorer outcomes. However, the genes and molecules that influence the disease course of sporadic ALS have not been established. Homozygous deletion of the SMN2 gene was suggested to be a prognostic factor in patients with sporadic ALS20; however, a subsequent study showed that the SMN2 gene copy number had no effect on survival time in patients with ALS.21 Reduced expression of the KIFAP3 gene has also been shown to increase survival time in patients with ALS22 ,23; however, subsequent reports suggested that the gene has no effect on survival.23–25 The EPHA4 gene expression in patients with ALS has shown to be correlated with age at disease onset. The influence of the EPHA4 gene on disease courses of ALS remains to be confirmed in prospective studies.26
In this study, we showed a significant association between a group of patients with sporadic ALS who showed a rapid decline in activities of daily living (ADL) and had seven SNPs related to decreased TTN expression. TTN encodes Titin, which is the largest human protein and is subject to extensive differential splicing. Titin spans the distance from the Z-line to the M-line in the striated muscle sarcomere and serves as a molecular ruler for the self-assembly of the sarcomere proteins during myofibrillogenesis, and also acts as a passive force generator.27–30 Mutations in TTN are found in various congenital myopathies, including limb-girdle muscular dystrophy,31 tibial muscular dystrophy32 and hereditary myopathy with early respiratory failure.33 In an animal model, the level of Titin in the skeletal muscles was preferentially reduced during long-term disuse, resulting in changes in the sarcomere structure.34 Based on these observations, decreased expression of TTN may influence the disease course in patients with ALS, possibly through decreased skeletal muscle function. Our results may suggest that if patients with ALS have minor homozygosity for the seven SNPs, those patients will show a rapid functional decline course with a probability of 0.359–0.382. If patients have major homozygosity or heterozygosity, the probability would be 0.082–0.107. We thus may be able to estimate whether the patients show the rapid decline course at certain probabilities by assessing these SNPs.
The genes and proteins that influence disease courses could be used to identify therapeutic targets or stratification factors for clinical trials. To date, there is only one drug—riluzole—that ameliorates the disease course of ALS. A number of candidate drugs for ALS therapy have been proven to show negative results in precisely designed clinical trials. Various reasons for these unfavourable results have been considered. The large diversity in the clinical courses of ALS is one of the reasons why detecting the defensive effects of candidate agents against disease progression may be difficult. The gene polymorphisms that influence the progression of symptoms in patients with ALS could be used to stratify the patients in a trial and might thereby increase the power of clinical trials. In addition, candidate drugs being tested in animal models of ALS are often given to the animals early in the disease course or even before disease onset; however, in human patients, treatment is often initiated months or years after disease onset, which might cause dissociation between the results from the animal models and clinical trials. Molecules that influence disease progression after ALS onset, such as Titin, might be used as targets to overcome this problem and develop therapeutic drugs. Our strategy and findings in this study will open the path to developing drugs to slow the ADL decline in patients with sporadic ALS.
The analyses conducted in the present study required hundreds of genome samples with long-term longitudinal clinical data. Therefore, replication analyses cannot be performed at present, which is a limitation of the present study. The outcome of the present study would be better confirmed through future studies with cohorts of patients with ALS in multiple ethnic populations. Another limitation of the present study is that the expression data was examined only in blood cells. Since the data in eQTL databases partly differ among the different tissues examined, the expression levels in various tissues including muscle should also be examined. Alternatively, we have recently established methods to obtain iPS cells from lymphocytes in a high-throughput manner with hundreds of samples, and we are currently generating cells of various tissue types, including muscles, motor neurons and glia cells. We will be able to analyse the expression profiles of the tissues obtained from the iPS cells of patients with the specific clinical phenotypes or genotypes as a future study.
Our strategy was to classify the disease course of a sporadic neurodegenerative disease and to identify genes associated with the different classified courses using GWAS. This strategy has the potential to identify modifiers of disease progression that can be used as a starting point to develop therapies for intractable neurodegenerative diseases such as ALS.
Acknowledgments
The authors thank all the patients with ALS who participated in this study. They also thank all the doctors and staff who participated in JaCALS.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
HaW, NA and AH contributed equally.
Twitter Follow Koji Abe at @Koji Abe
Collaborators The JaCALS members included Dr Tatsuhiko Yuasa (Kamagaya General Hospital); Dr Takahiro Kano (Hokkaido University); Dr Hidenao Sasaki (Hokkaido University); Dr Masaaki Kato (Tohoku University); Dr Takashi Imai (Miyagi National Hospital); Dr Tomohiko Ishihara (Niigata University); Dr Masatoyo Nishizawa (Niigata University); Dr Yukio Fujita (Gunma University); Dr Masaki Ikeda (Gunma University); Dr Kazumoto Shibuya (Chiba University); Dr Hideaki Hayashi (Tokyo Metropolitan Neurological Hospital); Dr Yuji Takahashi (National Center of Neurology and Psychiatry); Dr Miho Murata (National Center of Neurology and Psychiatry); Dr Hiroyuki Ishiura (The University of Tokyo); Dr Kotaro Ogaki (Juntendo University); Dr Nobutaka Hattori (Juntendo University); Dr Hiroyuki Tomiyama (Juntendo Tokyo Koto Geriatric Medical Center); Dr Hitoshi Aizawa (Tokyo Medical University); Dr Mie Nakamura (Tokyo National Hospital); Dr Yasuo Iwasaki (Toho University Omori Medical Center); Dr Takamura Nagasaka (University of Yamanashi); Dr Yoshihisa Takiyama (University of Yamanashi); Dr Tomokazu Obi (Shizuoka Institute of Epilepsy and Neurological Disorders); Dr Motoko Sakai (Suzuka National Hospital); Dr Masaaki Konagaya (Suzuka National Hospital); Dr Yu-ichi Noto (Kyoto Prefectural University); Dr Masanori Nakagawa (Kyoto Prefectural University); Dr Hirofumi Yamashita (Kyoto University); Dr Ryosuke Takahashi (Kyoto University); Dr Takuji Fujita (Takumikai Neurology Clinic); Dr Toru Yamashita (Okayama University); Dr Masanori Hiji (Vihara Hananosato Hospital); Dr Yasuhiro Watanabe (Tottori University); Dr Shintaro Hayashi (Kyushu University) and Dr Jun-ichi Kira (Kyushu University).
Contributors HaW contributed to drafting and revising the manuscript, acquisition, analysis and interpretation of the data, and execution of the research project. NA contributed to drafting and revising the manuscript, acquisition, analysis and interpretation of the data, execution of the research project, and development of the study design and concept. AH and MasahiNa contributed to drafting and revising the manuscript, analysis and interpretation of the data, and statistical analysis. RN contributed to acquisition and interpretation of the data, and execution of the research project. ShinI contributed to drafting and revising the manuscript, qRT-PCR experiments and data acquisition. AI, ShiroI and MKu contributed to typing of SNPs, revising the manuscript and interpretation of the data. DY contributed to acquisition and interpretation of the data and execution of the research project. HiW and MI contributed to acquisition and interpretation of the data, execution of the research project, and development of the study design and concept. MKa, YuI, KK, AT, IA, KA, KM, MO, OK, KoiO, SK, KH, TaI, AK and KN contributed to revising the manuscript, and acquisition and interpretation of the data. MMo, MA, ST and RK contributed to revising the manuscript, acquisition and interpretation of data, and serving on the JaCALS steering committee. GS contributed to research project organisation and execution, drafting and revising the manuscript, interpretation of the data, development of the study design and concept, and serving on the JaCALS steering committee. TY contributed to serving on the JaCALS steering committee. TK, HS, MKato, ToI, MaNi, FY, MaI, KS, AK, HH, YuT, MMu, HI, KotO, NH, HT, HitA, MiN, YaI, TN, YoT, OT, JS, MS, MKo, YN, MasanNa, HY, RT, TF, TY, MH, YW, SH, JK and IN contributed to acquisition of the data.
Funding This work was supported by a grant (15Aek0109071h0002) from Japan Agency for Medical Research and development (AMED), Health and Labour Sciences Research grant (H26-086) and Grants-in Aid (25461277) for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan. A section of this study is the result of the ‘Integrated Research on Neuropsychiatric Disorders’ study, which was carried out under the Strategic Research Program for Brain Sciences by the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Competing interests GS received grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan, during the conduct of the study.
Ethics approval The ethics committees of all the participating institutions.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Neurodegeneration