Article Text

Abstract
Multiple system atrophy (MSA) is a progressive neurodegenerative disorder characterised by a variable combination of autonomic failure, levodopa-unresponsive parkinsonism, cerebellar ataxia and pyramidal symptoms. The pathological hallmark is the oligodendrocytic glial cytoplasmic inclusion (GCI) consisting of α-synuclein; therefore, MSA is included in the category of α-synucleinopathies. MSA has been divided into two clinicopathological subtypes: MSA with predominant parkinsonism and MSA with predominant cerebellar ataxia, which generally correlate with striatonigral degeneration and olivopontocerebellar atrophy, respectively. It is increasingly recognised, however, that clinical and pathological features of MSA are broader than previously considered.
In this review, we aim to describe recent advances in neuropathology of MSA from a review of the literature and from information derived from review of nearly 200 definite MSA cases in the Mayo Clinic Brain Bank. In light of these new neuropathological findings, GCIs and neuronal cytoplasmic inclusions play an important role in clinicopathological correlates of MSA. We also focus on clinical diagnostic accuracy and differential diagnosis of MSA as well as candidate biomarkers. We also review some controversial topics in MSA. Cognitive impairment, which has been a non-supporting feature of MSA, is considered from both clinical and pathological perspectives. The cellular origin of α-synuclein in GCI and a ‘prion hypothesis’ are discussed. Finally, completed and ongoing clinical trials targeting disease modification, including immunotherapy, are summarised.
- multisystem atrophy
- neuropathology
- clinical neurology
- movement disorders
- cognition
Statistics from Altmetric.com
Introduction
Multiple system atrophy (MSA) is a progressive neurodegenerative disorder characterised by a variable combination of autonomic failure, levodopa-unresponsive parkinsonism, cerebellar ataxia and pyramidal symptoms.1 2 MSA was previously considered three distinct diseases: striatonigral degeneration (SND), olivopontocerebellar atrophy (OPCA) and Shy-Drager syndrome. Graham and Oppenheimer coined the term MSA to encompass the three entities.3 The discovery of glial cytoplasmic inclusion (GCI) in all three supports the unified concept of MSA.4 The current consensus criteria for the diagnosis of MSA define three degrees of certainty: definite, probable and possible.2 A diagnosis of definite MSA requires pathological confirmation.2 Autonomic failure, as exemplified by urinary incontinence or orthostatic hypotension, is a sine qua non for clinical diagnosing MSA.2 Depending upon the predominant motor symptom, MSA can be classified as two clinical phenotypes: MSA with predominant cerebellar ataxia (MSA-C) and MSA with predominant parkinsonism (MSA-P).2 Other than autonomic failure and motor symptoms, non-motor symptoms, including stridor, rapid eye movement sleep behaviour disorder, pseudobulbar affect, and severe dysphonia and dysarthria, are often seen in MSA.2 At present, there are no curative treatments for MSA; symptomatic treatment is, therefore, important.5
Neuropathology
Macroscopic findings
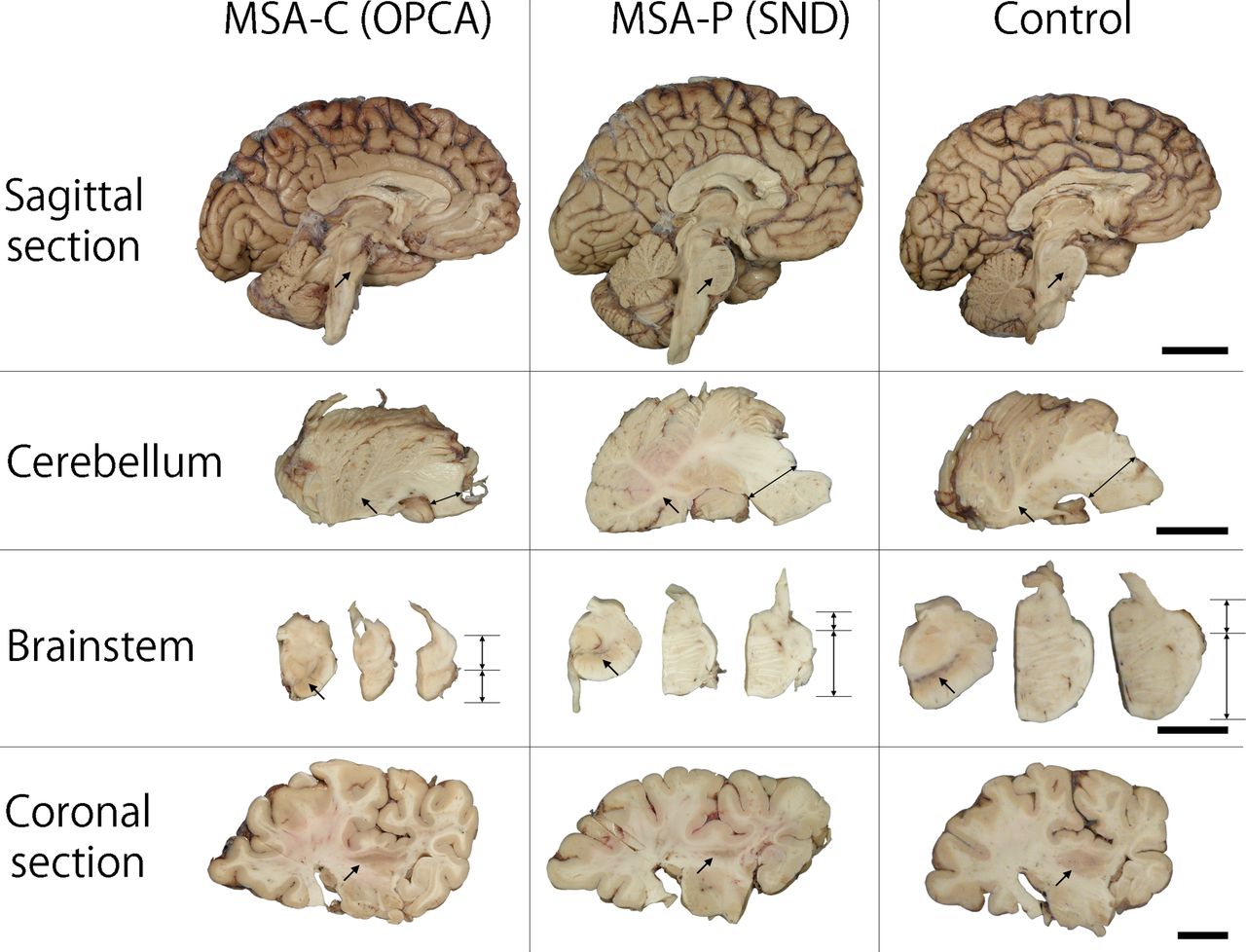
Macroscopically, a variable degree of brain atrophy is observed in cerebellar white matter, middle cerebellar peduncle, pontine base, medulla and posterolateral putamen, although the overall brain weight is generally within normal range. The neocortex and limbic structures are usually intact, except for atypical MSA (described in the Atypical MSA section) or MSA with other concomitant neurodegenerative conditions, such as Alzheimer disease (AD). Loss of pigment in the ventrolateral substantia nigra is often observed, especially, but not exclusively, in MSA-P. This finding is commonly seen in other parkinsonian disorders, including Parkinson’s disease (PD), dementia with Lewy bodies (DLB) and progressive supranuclear palsy (PSP). Discoloration and atrophy of the posterolateral putamen are specific features for MSA, especially MSA-P. Representative macroscopic findings in OPCA and SND compared with a neurologically normal brain are shown in figure 1.

Representative macroscopic findings of two pathological subtypes of MSA: OPCA and SND. In sagittal sections, significant atrophy of cerebellum and pons (arrow) is observed in OPCA. Atrophy of cerebellar white matter (arrow) and middle cerebellar peduncle (double-headed arrow) is observed in the section of cerebellum in OPCA. Grey-white matter differentiation is less visible in OPCA than in SND or control brain. Brainstem sections show pigment in the substantia nigra (arrow) and significant atrophy in the pontine base in OPCA. Note that the ratio of pontine tegmentum to pontine base is almost 1:1 in OPCA (double-headed arrows), compared with 4–5:1 in SND or control. Slight atrophy of the posterior putamen (arrow) is observed in the coronal section in OPCA. By contrast, the size of cerebellum and pons (arrow) is well preserved in SND. Cerebellar white matter (arrow) and middle cerebellar peduncle are also free of atrophy. Significant pigment loss in the substantia nigra (arrow) and severe atrophy of the posterior putamen (arrow) are observed. Bars: 4 cm in sagittal section, 2 cm in cerebellum and coronal section, 1.5 cm in brainstem. MSA, multiple system atrophy; MSA-C, MSA with predominant cerebellar ataxia; MSA-P, MSA with predominant parkinsonism; OPCA, olivopontocerebellar atrophy; SND, striatonigral degeneration.
Macroscopic findings are useful in differentiating MSA from other atypical parkinsonian disorders. Unlike PSP, atrophy of subthalamic nucleus, midbrain, cerebellar dentate nucleus and superior cerebellar peduncle are not observed in MSA. The absence of focal atrophy in the superior frontal and motor cortices differs from corticobasal degeneration.
Histopathological findings
System-specific neuronal loss and astrogliosis are observed in both striatonigral and olivopontocerebellar systems, regardless of the pathological subtypes (figure 2).6 The degree of neurodegeneration tends to correlate with disease duration and clinical phenotype.7 The severity of neuronal loss and astrogliosis in the putamen, globus pallidus and substantia nigra correlates with akinesia, while neuronal loss in the inferior olivary nucleus, pontine nucleus and cerebellum is associated with limb and gait ataxia.1 Myelin degeneration is also observed in affected regions, such as middle cerebellar peduncle, pons, caudate nucleus, putamen, globus pallidus and even in areas where GCIs were not detected (figure 2H).8 Excessive iron accumulation is observed in MSA with a different pattern compared with PD.9 An autopsy study demonstrated iron accumulation in the basal ganglia (figure 2G, inset) and substantia nigra in MSA,10 although the significance of this finding remains unknown.
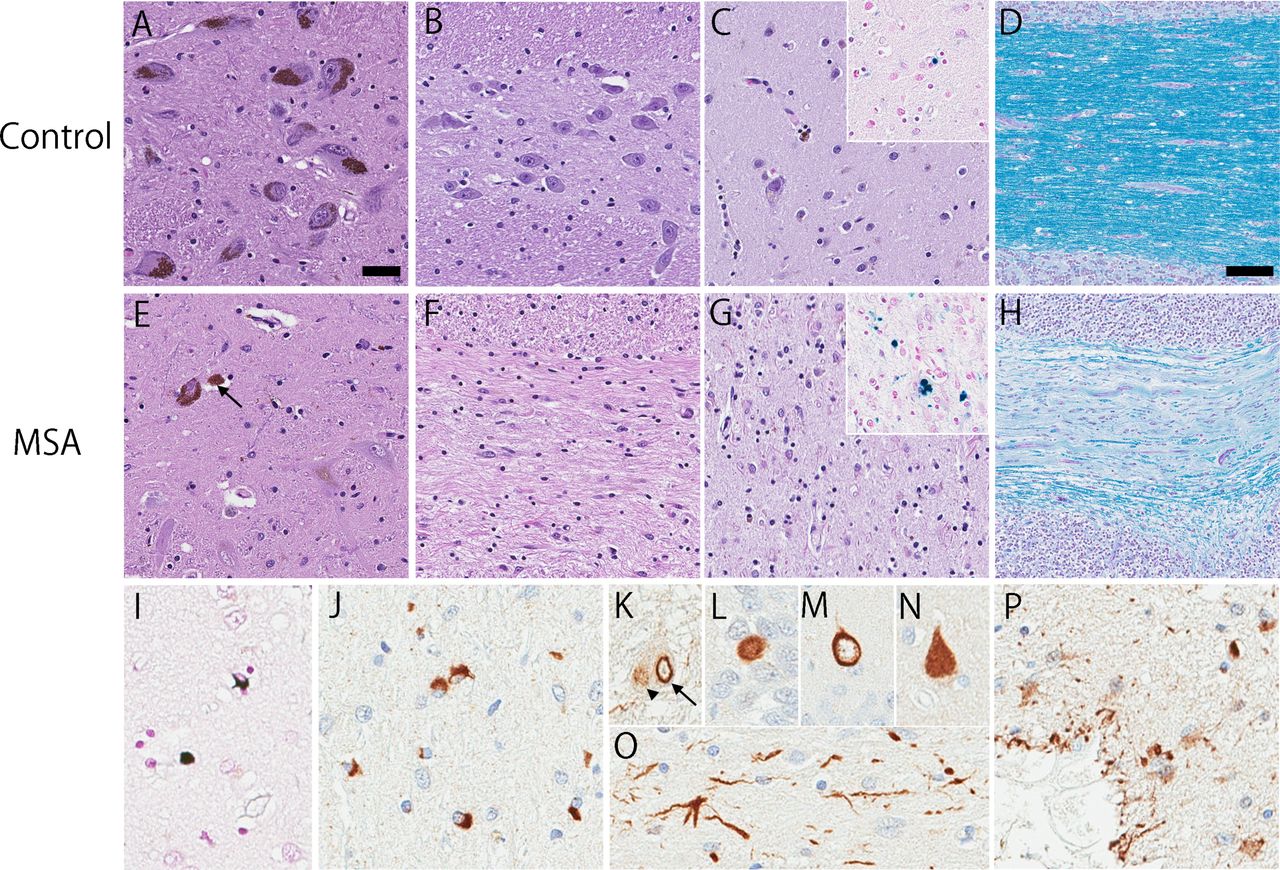
{kind=link}
{kind=link}
Neuronal loss, gliosis and demyelination in MSA are shown (E–H), compared with a neurologically normal control case (A–D). Prominent loss of neuromelanin containing neuron with extraneuronal melanin pigment (arrow) in the ventrolateral substantia nigra (E) is observed in MSA. The pontine nucleus shows significant neuronal loss and gliosis (F). Neuronal loss with gliosis and rarefaction of neuropil is observed in the lateral putamen (G). Prussian blue stain shows iron deposits in the putamen which are more frequent in MSA than in control case (inset, C and G). Luxol fast blue stain shows significant myelin pallor and demyelination in the cerebellar white matter (H). Glial cytoplasmic inclusions are argyrophilic inclusion oligodendrocytes (I) and are immunohistochemically positive for α-synuclein (J). Neuronal inclusions are commonly seen in the pontine base, including intranuclear inclusion (arrow in K), cytoplasmic inclusion (arrowhead in K) and dystrophic neurites (O). Neuronal cytoplasmic inclusions are also observed in the hippocampus in variable morphology, such as Pick body-like (L), ring-shaped (M) and neurofibrillary tangle-like inclusions (N). Subpial and perivascular astrocytes show α-synuclein immunoreactivity in a subset of MSA cases (P). Bars: 20 µm in A, 100 µm in D. All images are of equal magnification except D and H. A–C, E–G: H&E stain; insets in C and G: Prussian blue stain; D and H: Luxol fast blue stain; I: Gallyas silver stain; J–P: Immunohistochemistry with anti-α-synuclein antibody. MSA, multiple system atrophy.
Glial and neuronal α-synuclein pathologies
The presence of GCIs, argyrophilic inclusions in oligodendrocytes, is specific to MSA (figure 2I,J)4; therefore, widespread and abundant GCIs in association with neurodegenerative changes in striatonigral or olivopontocerebellar structures are criteria for definite neuropathological diagnosis of MSA.2 11 The main component of GCIs is α-synuclein; therefore, MSA is included in the category of α-synucleinopathies, along with PD and DLB.12 GCIs are found throughout the brain, with predilection for basal ganglia, substantia nigra, pons, inferior olivary nucleus and cerebellar white matter.6 13 The highest density of GCIs is in the basal ganglia, especially in the pencil fibres of Wilson in the putamen.13 GCIs can be detected in areas that do not show obvious neuronal loss; although neurodegeneration in the neocortex and limbic structures is usually minimal, the motor cortex and supplementary motor cortex often have many GCIs.13 14 The density of GCIs correlates with both neuronal loss and disease duration.6 13 15 16 These findings indicate that GCI likely plays a critical role in neurodegeneration in MSA, rather than a secondary consequence.
Neuronal cytoplasmic inclusions (NCIs) (figure 2K–N), neuronal intranuclear inclusions (figure 2K) and dystrophic neurites (figure 2O) are detected in most cases in the putamen, substantia nigra, pontine nuclei, inferior olivary nuclei, anterior cingulate cortex, amygdala, hippocampus, entorhinal cortex and hypothalamus.17 18 NCIs are heterogeneous in morphology, including Pick body-like (figure 2L), ring-shaped (figure 2M) and neurofibrillary tangle (NFT)-like inclusions (figure 2N). Compared with GCIs, the pathological significance of NCIs had not been the focus of much research in the past; however, recent studies suggest that NCIs may impact cognitive function (described in the Cognitive impairment in MSA section) and play a role in limbic system degeneration in atypical MSA (described in the Atypical MSA section).18 19
Recently, abnormally phosphorylated and aggregated α-synuclein has been recognised in subpial and periventricular astrocytes in MSA.20 21 Nakamura et al reported that these astrocytes were a specific feature of MSA, and that they correlated with disease duration.20 Subsequently, Koga et al found α-synuclein-positive astrocytes in subpial and perivascular regions in both MSA (figure 2P) and Lewy body disease, suggesting this pathology is not a specific feature of MSA.21
Staging systems
A grading system for SND was proposed based on semiquantitative assessment of atrophy, neuronal loss, astrogliosis and presence of GCI.22 Neuronal loss confined to the substantia nigra pars compacta is grade 1; neuronal loss extending to the putamen is grade 2; and further involvement of the caudate and globus pallidus is grade 3.22 Subsequently, the grading system was extended by Jellinger et al for both SND and OPCA.23 Of 42 patients, 22 were assigned as MSA-P and 20 were MSA-C. It is of note that no patients displayed ‘pure’ OPCA pathology or more severe OPCA pathology than SND (ie, OPCA III+SND I/II).23 These clinicopathological subtypes of MSA correlated with initial symptoms and clinical features of both types.23 Ozawa et al described pathological subtypes of MSA in two large series from UK and Japan using a simple grading system.16 Each case of SND and OPCA was divided into three grades based on semiquantitative assessment of neuronal loss in regions of interest: putamen, globus pallidus and substantia nigra for SND; pontine nuclei, cerebellar hemisphere and vermis, inferior olivary nucleus, and substantia nigra for OPCA.16 This classification showed significant clinicopathological correlation; the SND phenotype demonstrated more severe bradykinesia, and the OPCA phenotype showed more frequent cerebellar signs. In contrast to the study by Jellinger et al, no patients showed ‘pure’ SND or ‘pure’ OPCA in studies by Ozawa et al.16 23
Racial differences
The frequency of clinical phenotypes among different populations has been noted repeatedly in the literature. MSA-P is the predominant phenotype in Caucasian populations, whereas MSA-C is more frequent in Japanese populations.24–26 A North American study found that 60% of the patients had MSA-P and 13% had MSA-C.24 By contrast, 67%–84% of patients had MSA-C and 16%–33% had MSA-P in the studies in Japan.25 26 As expected, the differences in proportion of clinical phenotypes correlate with underlying pathological subtypes. Ozawa et al demonstrated different frequency of pathological subtypes between cohorts from UK and from Japan. A UK-based study showed that the frequency of mixed cases (equally severe in SND and OPCA regions) was 49%, followed by SND cases (34%) and OPCA cases (17%).16 In contrast, a Japan-based study showed that the frequency of mixed cases was 42%, followed by OPCA (40%) and SND (18%).27 The difference in frequency of pathological and clinical phenotypes among different populations suggests that genetic or environmental factors, or both, play a role in the pathogenesis of MSA, although almost all MSA cases are sporadic.
Minimal change MSA
Almost all MSA cases display neuronal loss in both striatonigral and olivopontocerebellar structures,16 27 with only 11 of 42 cases studied by Jellinger et al 23 assigned to the category of pure SND. Pure MSA cases may be valuable clues to elucidate the pathology in early stages of disease. The term ‘minimal change’ MSA was coined to describe two patients with clinically diagnosed MSA-P who had neuronal loss confined to the substantia nigra and locus coeruleus.28 Nevertheless, GCIs were observed throughout striatonigral and olivopontocerebellar structures. This finding suggests that GCI formation may precede neuronal loss. Minimal change variant has also been reported in a patient with MSA-C.29 Mild neuronal loss was restricted to the anterior vermis and inferior olivary nucleus, while GCIs were abundant in the pontine nuclei, middle cerebellar peduncle and cerebellar white matter.29 GCIs were also observed in the cerebral and spinal white matter, to a lesser extent.29
Neurologically normal individuals are rarely found to have GCIs at autopsy as a coincidental or incidental finding.30 31 Parkkinen et al screened 1800 brains with immunohistochemistry for α-synuclein and found one individual without any neurological symptoms who had abundant GCIs in the brainstem.30 Mild neuronal loss was confined to the substantia nigra and dorsal motor nucleus of vagus. Fujishiro et al screened two autopsy series with α-synuclein immunohistochemistry and identified two individuals with widespread GCIs without clinical symptoms of MSA (1 of 241 in the first series; 1 of 125 in the second series).31 Neuronal loss and gliosis were found in the ventrolateral substantia nigra in case 1 and case 2. The latter also showed neuronal loss in the dorsolateral putamen. Because all three cases in the two studies did not have clinical symptoms suggestive of MSA, the term incidental or preclinical MSA was used for these cases. As with minimal change MSA-P, neuronal loss was observed in the substantia nigra, suggesting this region might be the first affected in MSA, especially MSA-P.
Atypical MSA
Because MSA was conventionally divided into two pathological subtypes (ie, SND and OPCA), only striatonigral and olivopontocerebellar regions were systematically assessed in these grading systems; the limbic system and neocortex have been infrequently evaluated in MSA. Recently, a novel subtype of MSA, atypical MSA, also known as frontotemporal lobar degeneration (FTLD)-synuclein, has been introduced by Aoki and colleagues.19 The authors described four patients who clinically presented with frontotemporal dementia (FTD) syndromes, including corticobasal syndrome, progressive non-fluent aphasia and behavioural variant FTD.19 All had frontotemporal atrophy and severe α-synuclein neuronal pathology in limbic and neocortical regions.19 Rohan and colleagues also reported two patients with atypical MSA who had features similar to those reported by Aoki et al.32 Atypical MSA cases have also been reported from Japan. Of note, there are some differences in clinical presentations between atypical MSA in Western countries (ie, the USA and Europe) and Japan. Patients with atypical MSA in Japan had a younger age at onset and longer disease duration than those in Western countries.19 None of the patients had an antemortem diagnosis of MSA in Western countries (n=7), while at least three of five Japanese patients were diagnosed with MSA.19 The more severe brainstem and cerebellar involvement in Japanese patients may have led to recognition of MSA during life.
Concomitant pathology
Like other neurodegenerative diseases, ageing is one of the risk factors for MSA. Therefore, neuropathological hallmarks of other neurodegenerative diseases sometimes coexist with MSA. AD pathology is one of the most common pathologies in elderly individuals; however, concomitant AD pathology in MSA is less frequent than in age-matched controls.33 In an MSA series from the Mayo Clinic Brain Bank (n=102), only four cases (4%) had concurrent pathological AD.34
In a large series of MSA cases from the Queen Square Brain Bank, Lewy bodies, the pathological hallmark of PD and DLB, were found in 11% of cases (10 of 94).16 Similarly, 10% of MSA cases (10 of 102) from the Mayo Clinic Brain Bank had Lewy bodies.34 Intriguingly, Lewy body pathology was not observed in a series of 50 Japanese MSA cases.27 This discrepancy may be due to the genetic or environmental differences between Western countries and Japan.
PSP is an atypical parkinsonian disorder that sometimes clinically resembles MSA. Pathologically, PSP is a four-repeat tauopathy characterised by tau accumulation in both neurons and glia-forming NFTs, neuropil threads, tufted astrocytes and oligodendroglial coiled bodies. The rare coexistence of MSA and PSP has been reported in four individuals in the literature.35 Argyrophilic grain disease (AGD) is another four-repeat tauopathy, characterised by small dot or grain-shaped dendritic inclusions, so-called argyrophilic grains, in the limbic regions in elderly individuals.36 AGD frequently coexists with other neurodegenerative conditions. Wakabayashi et al reported that argyrophilic grains were found in the limbic system in 5 of 26 patients with MSA (19%), and 2 of them developed mild dementia.37 Chronic traumatic encephalopathy (CTE) is a recently recognised tauopathy associated with repetitive traumatic brain injury, pathologically characterised by NFT and thorn-shaped astrocyte in perivascular foci, often at the depths of cerebral sulci of convexity frontal and temporal cortices. Of the 139 MSA cases from the Mayo Clinic Brain Bank, 8 (6%) had CTE pathology.38 All eight patients with MSA with CTE pathology were male and four of them had a documented history of exposure to contact sports. Repetitive falls due to MSA may also contribute to this pathology, but further investigation is necessary to support this hypothesis. Ageing-related tau astrogliopathy (ARTAG) is a term describing a morphological spectrum of tau-positive astroglial pathology mainly observed in elderly individuals.39 In the same series of MSA screened for CTE pathology, 10 (7%) had ARTAG.38 As expected, age at death was older in MSA with ARTAG than in cases without ARTAG.38
The TAR DNA-binding protein of 43 kDa (TDP-43), originally identified as a major component of ubiquitin-positive/tau-negative inclusions in FTLD and motor neuron disease, was shown to occur rarely in MSA.40 Geser et al found only 14% of MSA cases (4 of 29) with at least a mild level of TDP-43 pathology.40 In a series of 148 MSA cases from the Mayo Clinic Brain Bank, only 10 (7%) had TDP-43 pathology in the amygdala (Koga et al, unpublished data). Interestingly, the average of age at death was significantly older in TDP-43-positive MSA than in TDP-43-negative patients (77 years vs 66 years, p=0.025). This finding suggests that the TDP-43 pathology in MSA is likely to be an age-related pathology rather than a disease-specific change.
Differential diagnosis
Diagnostic accuracy and differential diagnosis
Due to the heterogeneity of clinical phenotype and lack of specific biomarkers, it is challenging to make a correct antemortem diagnosis of MSA.41 Since a definite diagnosis is confirmed at autopsy, discrepancies between clinical and pathological diagnoses are inevitable. Osaki et al assessed the accuracy of a clinical diagnosis of MSA; 86% (51 of 59) of patients with clinically diagnosed MSA were confirmed at autopsy.42 A recent study reported 90% diagnostic accuracy, although the sample size was relatively small (9 of 10).43 In contrast, a large series of MSA from Mayo Clinic Brain Bank reported an unexpectedly low diagnostic accuracy. Of 134 patients with clinically diagnosed MSA, only 83 (62%) had definite MSA at autopsy.41 Patients with DLB, PD and PSP were often misdiagnosed with MSA. Autonomic failure was the leading cause of misdiagnosis in DLB and PD, and cerebellar ataxia was the leading cause of misdiagnosis in PSP.41 PSP with predominant cerebellar ataxia, a rare clinical phenotype of PSP, is often misdiagnosed as MSA-C because of similarities in disease presentation.44 45 Corticobasal degeneration, normal pressure hydrocephalus and vascular parkinsonism can also mimic MSA.41 46
Although MSA is a sporadic disease, genetic disorders that present with progressive adult-onset cerebellar ataxia are included in the differential diagnoses of MSA-C. Apparently, sporadic spinocerebellar ataxia (SCA) with autonomic failure can masquerade as MSA-C. Indeed, a recent study reported that 7% of patients with clinically diagnosed MSA had mutations in SCA genes.47 Fragile X tremor-ataxia syndrome can be misdiagnosed as MSA because of the presence of ataxia and parkinsonism. Interestingly, among the patients with probable MSA-C, 4% (3 of 76) carried FMR1 premutations.48 The rare cerebello-brainstem dominant form of X-linked adrenoleukodystrophy has also been reported to mimic MSA-C.49
Fluid biomarkers
A number of studies have sought candidate biomarkers using blood and cerebrospinal fluid (CSF), but no reliable biomarkers are currently available for diagnosis of MSA (reviewed in ref 50). Total α-synuclein levels in CSF have been often examined, but with inconsistent results. Of nine studies using antemortem CSF, six revealed decreased total α-synuclein levels in patients with MSA compared with control subjects, while three other studies did not show difference between the two groups (table 1). Importantly, of the six studies, only one demonstrated differences between MSA and PD, suggesting limited usefulness for differential diagnosis among patients with parkinsonism. A major problem of total α-synuclein levels in CSF is great variability among CSF levels in each study. This could be explained by possible blood contamination and different methods used to detect total α-synuclein levels in CSF.50
Select candidate fluid biomarkers for MSA
Besides α-synuclein, various proteins (eg, DJ-1 and tau), catecholamines (eg, dopamine) and their metabolites, and neuroinflammatory cytokines have been examined (table 1).50–55 Although no single biomarker is sensitive and specific, combinations of each biomarker could be more useful.50 A combination of DJ-1 and total tau showed high sensitivity (82%) and specificity (81%) for differentiating MSA from PD.56 A caveat, however, is that most studies are based on patients with clinically diagnosed MSA, so patient populations are inevitably heterogeneous due to variable accuracy of clinical diagnosis. Validation studies using patients with autopsy-confirmed MSA are needed to establish fluid biomarkers for diagnosis.
Molecular and functional imaging
Conventional imaging modalities (ie, CT and MRI) and molecular and functional imaging have been used in the differential diagnosis of MSA (reviewed in ref 57). Hypometabolism, decreased activity of dopaminergic and cholinergic systems, and neuroinflammation have been visualised in brain regions vulnerable to MSA (table 2). It is expected that tau positron emission tomography (PET) imaging will improve the clinical diagnostic accuracy of MSA by excluding parkinsonian tauopathies that can masquerade as MSA; however, interpretation of tau PET should be done cautiously since some patients with MSA with severe GCI pathology might be false positives.58 59 A study on [18F]AV-1451 PET scan disclosed a significant retention in posterior putamen in four consecutive patients with MSA.58 [11C]PBB3 PET also showed increased binding in cortical and subcortical regions in a patient with MSA (table 2).59 Based on matched in vitro fluorescence and autoradiographic binding for PBB3 in the same brain regions, a subset of patients with MSA could be positive because of the high density of GCI in the putamen,60 even though the affinity of PBB3 to α-synuclein is 10–50 times less than that to tau.61 These tau ligands bind to β-pleated sheet secondary structures in tau filaments, which are also characterised by α-synuclein filaments. Future clinical studies will be needed to determine the usefulness of tau PET imaging for differential diagnosis of atypical parkinsonian disorders. Although it has yet to be reported, in vivo imaging of α-synuclein pathology could be a direct biomarker and a tool for drug development in α-synucleinopathies.
Molecular and functional imaging in MSA
Controversies
Cognitive impairment in MSA
Unlike other α-synucleinopathies such as PD and DLB, MSA has not been considered as a disease associated with significant cognitive impairment. According to consensus criteria, dementia (as defined by DSM-IV criteria) is a non-supporting feature for a clinical diagnosis of MSA2; however, a position statement by the Neuropsychology Task Force of the Movement Disorders Society MSA study group indicates that cognitive impairment may be an under-recognised feature in MSA (reviewed in ref 62). The frequency of cognitive impairment varies depending on the study design and sample size, but ranges from 14% to 37%.1 18 34 63 64 The pattern of cognitive impairment in MSA is similar to that in PSP, which is sometimes referred to as ‘subcortical dementia’, although less severe in MSA than in PSP.65 66 Frontal executive function is most frequently affected, while memory and visuospatial domains are less often impaired.34 62 65–68 Processing speed deficits have also been identified, but a caveat is that motor deficits in MSA can affect interpretation of this domain.69 70 The Frontal Assessment Battery and Initiation/Perseveration Subscale of the Dementia Rating Scale-II can be useful in detecting frontal executive dysfunction in MSA.66 The Montreal Cognitive Assessment was more sensitive than Mini-Mental State Examination (MMSE) in detecting cognitive impairment in MSA, but using a MMSE threshold of <27 increased its sensitivity, suggesting that this could be a practical screening tool for cognitive impairment in MSA.68 71 Several imaging studies have found that thinning in neocortices and volume reduction in subcortical structures were associated with cognitive impairment in MSA.72–74 A multicentre MRI study showed that cognitive impairment in MSA was associated with focal volume reduction in the left dorsolateral prefrontal cortex, which was considered secondary to underlying subcortical degeneration.74 These antemortem findings are valuable for suggesting regions that are associated with cognitive impairment, but autopsy-based correlates are needed to validate these results.
The underlying neuropathology of cognitive impairment in MSA is not understood. Asi et al reported no significant difference in GCI and NCI burdens in limbic or cortical regions in patients with MSA with and without cognitive impairment.75 Recent studies, however, have shown that NCIs rather than GCIs in the neocortex or limbic regions may impact cognitive impairment in MSA.18 34 63 Cykowski et al concluded that Lewy body-like neuronal inclusions in neocortex were associated with cognitive impairment.18 Homma et al reported that frequent globular NCIs in the medial temporal regions are a pathological feature of dementia in MSA.63 Our previous study also showed that greater burden of NCI in hippocampal dentate gyrus was associated with cognitive impairment in MSA.34 More recently, a patient with MSA-dementia who had numerous NCIs in the perirhinal region without hippocampal involvement has been reported.76 Nevertheless, a correlation between the domains or degree of cognitive impairment and respective functional neuroanatomical regions remains unclear. Further clinicopathological studies are necessary to elucidate pathological mechanisms of cognitive impairment in MSA.
Where is the cellular origin of α-synuclein in GCI?
Although it is incontestable that the formation of GCI is closely associated with the underlying pathology of MSA, the mechanism of GCI formation has not yet been elucidated. In vitro study demonstrated that p25α, an oligodendroglial-specific protein, stimulated α-synuclein aggregation in an oligodendroglial cell culture model.77 In MSA brains, relocation of p25α from the myelin sheath and nucleus to the cytoplasm has been considered to precede aggregation of α-synuclein78–80; however, the cellular origin of α-synuclein is controversial. α-Synuclein is a neuronal protein that localises predominantly to presynaptic terminals.81 Even though accumulation of α-synuclein in oligodendrocytes is the pathological hallmark in MSA, SNCA mRNA was not expressed in oligodendrocytes in MSA or control brains.82 83 Therefore, it has been hypothesised that α-synuclein in GCIs in MSA could be of neuronal origin and taken up by oligodendrocytes. Cell culture studies showed that oligodendrocytes had the ability to take up recombinant α-synuclein monomer or α-synuclein secreted from neurons overexpressing α-synuclein.84 85
On the other hand, findings that challenge this hypothesis have been reported.81 86 Asi and colleagues demonstrated SNCA mRNA expression in oligodendrocytes in MSA postmortem tissue using microdissection and quantitative PCR.86 Although not statistically significant, SNCA mRNA level may be increased in MSA compared with control brains.86 Subsequently, Djelloul and colleagues identified SNCA transcripts in oligodendrocyte lineage cells, including embryonic stem cell and induced pluripotent stem cell-derived oligodendrocytes from patients with MSA.81 This finding is in line with those of Asi’s study, but it remains to be determined whether the SNCA transcripts identified are reminiscent of basal expression in quiescent oligodendrocyte precursor cells. Furthermore, α-synuclein expression decreased during oligodendrocyte maturation.81 Taken together, SNCA transcripts identified in oligodendrocyte lineage cells may not be the origin of α-synuclein in GCIs in MSA.
Is MSA a prion disease?
Recent studies using cell culture and animal models have shed light on the possibility that α-synuclein can be propagated like a prion protein (reviewed in refs 87 and 88). Luk et al inoculated wild-type mice with synthetic mouse α-synuclein preformed fibrils (PFFs) into the striatum and found widespread α-synuclein pathology and dopaminergic neuronal loss in the substantia nigra pars compacta.89 In contrast, α-synuclein knockout mice inoculated with PFFs did not develop α-synuclein pathology.89 These findings suggested that endogenous α-synuclein was necessary to propagate α-synuclein pathology. Subsequently, Masuda-Suzukake et al demonstrated transmission of α-synuclein pathology from human brain samples.90 Intracerebral injections of a sarkosyl-insoluble α-synuclein homogenates from DLB brains induced hyperphosphorylated α-synuclein pathology in wild-type mice.90 Intriguingly, exogenous human α-synuclein disappeared a week after inoculation, while endogenous mouse α-synuclein was converted into a pathological form and accumulated in neurons through a prion-like mechanism at 3 months postinoculation.90 The authors also demonstrated that propagation of phosphorylated α-synuclein occurs along neural circuits and involves trans-synaptic transport.91 These results suggest that exogenous pathological α-synuclein can convert endogenous α-synuclein into a pathological form that can propagate in a neuron-to-neuron manner. Nevertheless, these inoculated wild-type mice did not develop motor deficits.
Homozygous transgenic mice expressing human α-synuclein with A53T mutation, termed TgM83+/+ mice, spontaneously develop motor deficits around 1 year of age, along with widespread α-synuclein pathology.92 Mougenot et al inoculated young asymptomatic TgM83+/+ mice with a brain homogenate from old TgM83+/+ mice with motor deficits.93 The inoculated mice developed characteristic motor clinical signs earlier than uninoculated TgM83+/+ mice or mice inoculated with a brain homogenate from young asymptomatic TgM83+/+ mice.93 The heterozygous TgM83+/− did not develop motor deficits spontaneously, but inoculation with a brain homogenate from old TgM83+/+ mice induced neurological dysfunction in about 210 days.94 The authors also demonstrated that brain homogenates from patients with MSA induced motor deficits and α-synuclein pathology.94 95 The TgM83+/− mice inoculated with brain homogenates from two patients with MSA developed progressive motor deficits about 100 days after inoculation. A subsequent study with brain homogenates from 14 patients with MSA and six patients with PD revealed that all MSA brain extracts transmitted to TgM83+/− mice had neurodegeneration, but none of the PD extracts had neurodegeneration.95 This finding suggests that α-synuclein in MSA may be distinct from that in PD in prion-like properties.
There are challenges to the hypothesis that MSA is a prion disease. First, endogenous wild-type α-synuclein is insufficient to propagate α-synuclein pathology; mutated α-synuclein is needed as a template. Wild-type mice inoculated with PFFs or brain homogenates from MSA brains did not develop overt motor deficits despite having α-synuclein pathology.89 90 Transgenic mice expressing wild-type α-synuclein also developed α-synuclein deposition at 6 months of age, but no motor signs were observed.96 Second, although the pathological hallmark of MSA is GCIs in oligodendrocytes, phosphorylated α-synuclein aggregates were not detected in oligodendrocytes in MSA-inoculated TgM83+/− mice.94 95 Similar studies using mice expressing human α-synuclein under an oligodendrocyte-specific promoter may be needed to provide evidence of oligodendrogliopathy consistent with MSA.
Translational and therapeutic approach
Ongoing and completed clinical trials targeting disease modification listed on ClinicalTrials.gov. are summarised in table 3.97 98 Unfortunately, most of clinical trials have failed to show positive results, probably because of small number of enrolled patients and inevitable involvement of non-MSA patient. Also, by the time the patient is symptomatic, it may be too late for disease-modifying intervention. The only clinical trial showing positive result is intra-arterial and intravenous injection of autologous mesenchymal stem cells (MSCs), which delayed disease progression as measured by unified MSA rating scale in patients with MSA-C.99 A phase I clinical trial of intrathecal administration of MSCs for patients with MSA conducted by Mayo Clinic is ongoing (NCT02315027).
Ongoing, completed and terminated clinical trials targeting disease modification for MSA
Targeting the ‘prion-like’ cell-to-cell propagation of α-synuclein, immunotherapy for α-synuclein is an appealing approach and has been studied in animal models of MSA.100 Active immunisation with AFFITOPE AFF1 successfully decreased accumulation of α-synuclein, and reduced demyelination and neurodegeneration in myelin basic protein (MBP)-α-synuclein transgenic mice, a model of MSA that expresses α-synuclein in oligodendrocytes.101 A phase I clinical trial with AFFITOPE vaccines (PD01A and PD03A) has been completed, and results not yet reported (NCT02270489). Passive immunisation against α-synuclein also demonstrated positive results in mouse model of α-synucleinopathies.102 A combination of a single-chain antibody (CD5-D5) and anti-inflammatory treatment (lenalidomide) ameliorated gliosis, α-synuclein accumulation and behavioural deficits in MBP-α-synuclein transgenic mice.103 Although no clinical trials with passive immunotherapy are ongoing in patients with MSA, a phase Ib study of PRX002, an antibody against α-synuclein, in healthy volunteers showed favourable safety, tolerability and pharmacokinetic profiles.104
Conclusions
It is increasingly recognised that clinical and pathological features of MSA are broader than previously considered. A subset of patients with MSA develops cognitive impairment that shares features with PSP. In rare cases, patients with atypical MSA present with FTD syndromes, which is at variance with the classical picture of SND, OPCA or Shy-Drager syndrome. In light of these new neuropathological findings, GCIs and NCIs must be considered to play an important role in clinical features of MSA. The relatively low clinical diagnostic accuracy is one of the obstacles in designing clinical studies of high quality in MSA. Potentially heterogeneous participants of studies may fail to show expected results in clinical trials or genetic research, especially if sample sizes are small. The development of reliable biomarkers, with α-synuclein PET being a promising candidate, may overcome this obstacle and facilitate translational research. Analysing clinical features of patients with pathologically confirmed MSA identifies additional red flags, which may lead to refinements in diagnostic criteria. While α-synuclein clearly is found in GCIs, how it gets there is controversial, and evidence that MSA extracts propagate disease in animal models needs to be grounded in relevance of these findings to humans. Development of cellular and animal models that recapitulate underlying pathology in MSA will solve issues related to this insoluble protein.
Acknowledgments
We would like to thank the patients and their families who donated brains to help further the scientific understanding of neurodegeneration. The authors would also like to acknowledge Linda Rousseau and Virginia Phillips for histological support, and Monica Castanedes-Casey in Mayo Clinic, Jacksonville for immunohistochemistry support.
References
Footnotes
Contributors SK and DWD contributed to the conception of the manuscript. SK contributed to the literature review, initial drafts of the manuscript, and preparing tables and figures. DWD contributed to the final editing of the manuscript.
Funding This research is supported by NIH grant P50 NS072187 and a Jaye F and Betty F Dyer Foundation Fellowship in progressive supranuclear palsy research.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Commissioned; externally peer reviewed.
Correction notice This article has been corrected since it was published Online First. Reference 69 has been corrected.