Article Text

Abstract
Background We evaluated the seroprevalence of myelin oligodendrocyte glycoprotein immunoglobulin G1 (MOG-IgG) and associated clinical features of patients from a large adult-dominant unselected cohort with mainly relapsing central nervous system (CNS) inflammatory diseases. We also investigate the clinical relevance of MOG-IgG through a longitudinal analysis of serological status over a 2-year follow-up period.
Methods Serum samples from 505 patients with CNS inflammatory diseases at the National Cancer Center were analysed using cell-based assays for MOG-IgG and aquaporin-4 immunoglobulin G (AQP4-IgG). MOG-IgG serostatus was longitudinally assessed in seropositive patients with available serum samples and at least 2 years follow-up.
Results Twenty-two of 505 (4.4%) patients with CNS inflammatory diseases were positive for MOG-IgG. Patients with MOG-IgG had neuromyelitis optica spectrum disorder (NMOSD, n=10), idiopathic AQP4-IgG-negative myelitis (n=4), idiopathic AQP4-IgG-negative optic neuritis (n=4), other demyelinating syndromes (n=3) and multiple sclerosis (n=1). No relapses were seen in patients when they became MOG-IgG seronegative, whereas a persistent positive serological status was observed in patients with clinical relapses despite immunotherapy.
Conclusions In a large adult-predominant unselected cohort of mainly relapsing CNS inflammatory diseases, we confirmed that NMOSD phenotype was most commonly observed in patients with MOG-IgG. A longitudinal analysis with 2-year follow-up suggested that persistence of MOG-IgG is associated with relapses.
Statistics from Altmetric.com
Introduction
Myelin oligodendrocyte glycoprotein (MOG) is a component of myelin proteins in the central nervous system (CNS).1 MOG is predominantly located in the outermost surface of myelin sheaths and is a biologically accessible antigenic target for circulating autoantibodies.1 The contribution of MOG and anti-MOG immunoglobulin G (MOG-IgG) in CNS demyelination has been studied using animal models immunised with MOG.2 3
In humans, the role of MOG-IgG is still emerging. Indeed, diverse clinical manifestations of patients with MOG-IgG have been described in inflammatory CNS diseases with benign to fulminant features, including monophasic or relapsing clinical courses.4–27 Additionally, the phenotype can include neuromyelitis optica spectrum disorder (NMOSD), acute disseminated encephalomyelitis (ADEM) and rarely multiple sclerosis (MS) in both the adult and paediatric population.4–27
The majority of previous studies that characterise patients with MOG-IgG have been performed in cohorts selected with specific disease or phenotype.7–24 More importantly, the clinical implication of MOG-IgG status for prognosis and treatment has not been fully elucidated. Understanding risk factors for relapses would allow more directed patient treatment; hence, in the present study, we evaluated the seroprevalence of MOG-IgG and clinical spectrum of associated disorders in a large unselected cohort of inflammatory CNS diseases. We also investigated the clinical relevance of MOG-IgG in relapses through the longitudinal analysis of serological status.
Methods
Patients
Between 2005 and 2016, at the Department of Neurology of the National Cancer Center (NCC) in Korea, 505 consecutive patients with suspected inflammatory demyelinating CNS diseases who had available serum samples were enrolled; 199 had paired cerebrospinal fluid (CSF) available. Serum and CSF samples from all participants were stored at −80°C before analysis. The enrolled patients presented with the following diagnoses determined at their last visit: 243 with NMOSD, 130 with MS, 104 with idiopathic aquaporin-4 immunoglobulin G (AQP4-IgG)-negative myelitis, 9 with idiopathic AQP4-IgG-negative optic neuritis and 19 with other demyelinating syndromes that did not fit either NMOSD or MS. Diagnoses of NMOSD or MS were based on the 2015 International Panel for NMO Diagnosis (IPND) criteria or the 2010 McDonald criteria, respectively.28 29 Patients with idiopathic AQP4-IgG-negative myelitis or optic neuritis had the lesion(s) confined to spinal cord or optic nerve, respectively. The clinical, laboratory and radiological data of these patients were reviewed retrospectively.
The Institutional Review Board of NCC approved the present study and written informed consent was obtained from all participants.
MOG-IgG cell-based assays
Serum samples were evaluated by detecting IgG1 antibodies targeting full-length MOG using a cell-based assay (CBA) at Oxford University, UK, as previously described.30 AQP4-IgG was examined in-house at the NCC and/or commercial CBA (Euroimmun, Luebeck, AG, Germany).31 32 AQP4-IgG was evaluated on at least two independent tests and by two independent investigators (GYK and YSK). To confirm the simultaneous presence of both MOG-IgG and AQP4-IgG, MOG-IgG-positive samples were re-examined for AQP4-IgG status at Oxford University.33 The testing investigators were blind to the diagnosis and clinical data for the both MOG-IgG and AQP-IgG assays. In patients positive for MOG-IgG who had available serum samples with at least 2 years of follow-up, longitudinal analysis of MOG-IgG was subsequently performed.
Results
Clinical characteristics of patients with MOG-IgG
MOG-IgG was positive in 22 of 505 patients (4.4%) and AQP4-IgG was positive in 212 of 505 patients (42%). One of 505 patients (0.2%) was positive for both antibodies. Of the 22 patients with MOG-IgG, 21 patients had paired serum and CSF samples. Of them, three patients showed MOG-IgG positivity in both serum and CSF, while 18 patients were positive only in serum.
The demographics and clinical characteristics of 22 patients positive for MOG-IgG are shown in table 1. The median age at the onset was 30 years with a range of 4 to 50 years, and 14 patients were female. The median follow-up duration was 63 months (range of 7–200 months). Seventeen had relapsing clinical courses (77.3%) within the median follow-up duration of 71 months (range of 23–200 months), while only five patients had a monophasic clinical course (median follow-up duration of 34 months (range of 7–113 months)). At last follow-up, the Expanded Disability Status Scale (EDSS) score was highly variable, ranging from 0 to 7.0 (median 2.0), and the median number of attacks was 3 (range of 1–16). Twenty-two patients positive for MOG had the following diagnoses at the last visit: 10 with NMOSD, four with idiopathic AQP4-IgG-negative myelitis, four with idiopathic AQP4-IgG-negative optic neuritis, three with other demyelinating syndromes and one with MS. The most common initial clinical phenotype was optic neuritis (36%) followed by myelitis (27%), brain involvement (23%) and multifocal involvement (14%).
Demographics and clinical features of 22 patients with MOG-IgG
The serological status of MOG-IgG and AQP4-IgG according to the clinical diagnosis at the last follow-up is shown in figure 1. Of the 243 patients with NMOSD who fulfilled the IPND criteria, 212 (87.2%) were positive for AQP4-IgG, 10 (4.1%) were positive for MOG-IgG, 22 (9.1%) were double negative and 1 (0.4%) was double positive for both AQP4-IgG and MOG-IgG. The double-positive patient exhibited typical NMOSD features characterised by recurrent episodes of optic neuritis and longitudinally extensive transverse myelitis. One (0.8%) of 130 patients with MS was MOG-IgG positive. The patient with MS positive for MOG-IgG demonstrated the clinical and radiologic features (figure 2) consistent with relapsing MS, and responded well to interferon-ß therapy. Additionally, she revealed CSF-restricted oligoclonal bands and a new enhancing lesion on brain MRI was observed between clinical relapses. Four of 104 (3.8%) patients with idiopathic AQP4-IgG-negative myelitis, 4 of 9 (44.4%) patients with idiopathic AQP4-IgG-negative optic neuritis and 3 of 19 (15.8%) patients with other demyelinating syndromes were positive for MOG-IgG. The detailed clinical features of individual patients with MOG-IgG are shown in table 2.

Patient diagnosis at last follow-up with serological status of MOG-IgG and AQP4-IgG. AQP4, aquaporin-4 antibody; MOG, myelin oligodendrocyte glycoprotein antibody; MS, multiple sclerosis; NMOSD, neuromyelitis optica spectrum disorder.
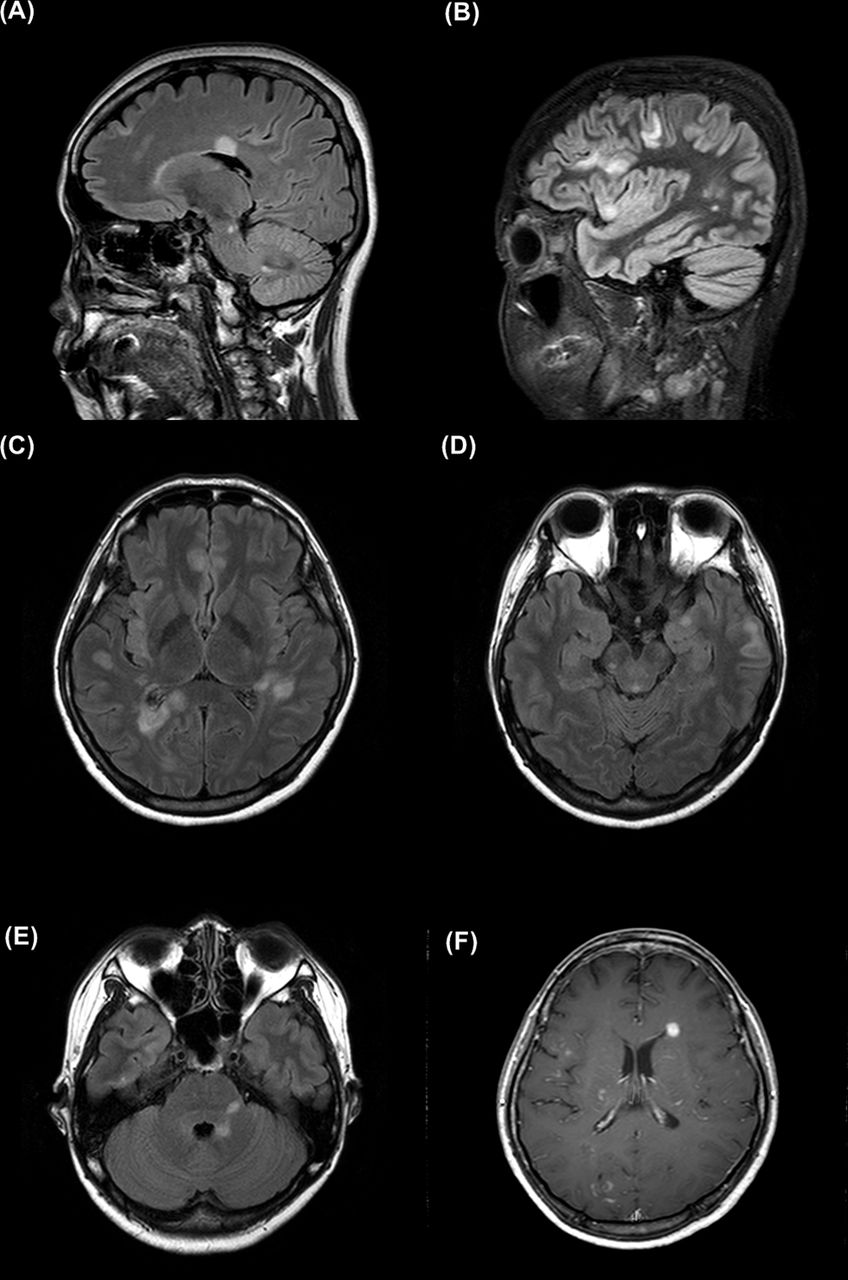
Representative MRI findings of the patient with multiple sclerosis juxtacortical (A,B), periventricular (A,C,F), infratentorial (A,D,E), inferior temporal (D), U-fibre (A,B) and nodular enhancing periventricular (F) lesions. These images were taken at 44 (A), 45 (B–E) and 47 (G) months after onset, and are highlighted on figure 3G as B1, B2 and B3.
Clinical characteristics of 22 individual patients with MOG-IgG
Longitudinal analysis of patients with MOG-IgG
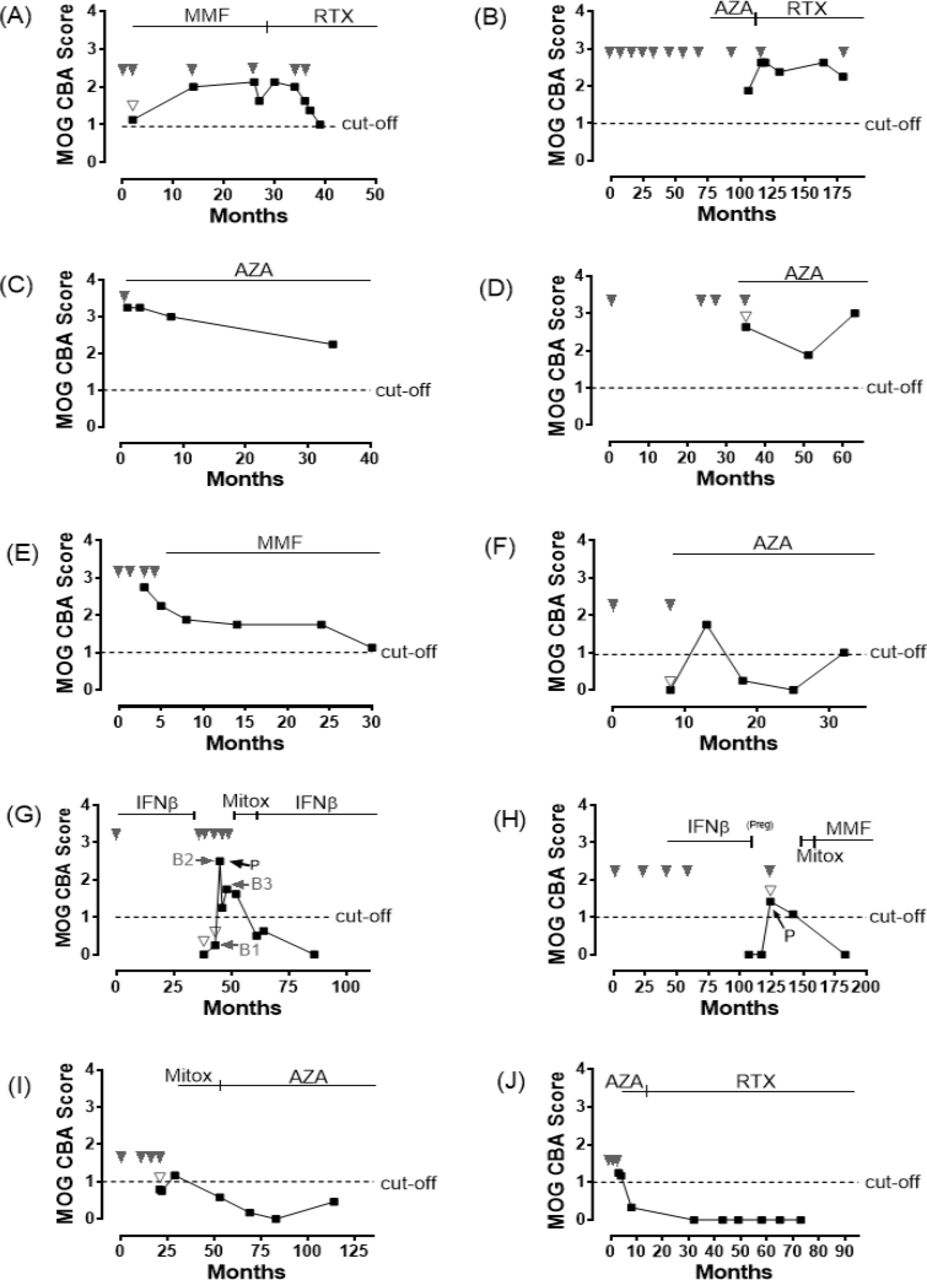
Longitudinal serum samples were obtained in 10 of 22 MOG-IgG seropositive patients with median follow-up of 39 months (range 27–92), and all 10 patients were treated with maintenance immunotherapy including immunosuppressive and immune-modulating therapy. Longitudinal analysis of 10 patients with MOG-IgG was demonstrated in figure 3 (patients 1–10 in table 2 are shown as A–J, respectively) and the detailed clinical characteristics of these 10 patients are shown in table 2. Of 10 patients, 5 (50%, figure 3A–E) showed persistent MOG-IgG positivity during a median of 33 months follow-up (range of 28–73 months), and one (figure 3F) patient became seronegative but returned to seropositive status during 28 months of follow-up. Four patients (40%, figure 3G–J) became MOG-IgG negative during a median of 73 months follow-up (range of 48–100 months).

{kind=link}
{kind=link}
{kind=link}
Longitudinal analysis of 10 patients with myelin oligodendrocyte glycoprotein immunoglobulin G (MOG-IgG). Persistent MOG-IgG positivity was observed in six patients and four patients showed negative serological conversion of MOG-IgG (black squares represented serial MOG antibody status, downward solid arrowheads represented clinical relapse, downward empty arrowheads represented serum samples which were obtained after steroid pulse therapy, upward arrows represented plasmapheresis and horizontal arrows showed time points of the representative MRI taken in figure 2, cut-off value for positivity: dotted line (score 1); the MOG-IgG CBA is scored visually in a semiquantitative manner: a CBA score of less than 1 is negative, a score of 1–1.5 is considered borderline/low positive and above 1.5 is positive. AZA, azathioprine; CBA, cell-based assay; IFNβ, interferon-beta; Mitox, mitoxantrone; MMF, mycophenolate mofetil; P, plasmapheresis; preg, pregnant; RTX, rituximab.
Of the 5 (50%) patients with a persistent MOG-IgG-positive status, two patients had recurrent attacks despite immunotherapy with rituximab (figure 3A,B). In contrast, no patient who became MOG-IgG seronegative showed clinical relapses after undergoing immunotherapy (figure 3G–J; table 2). Table 3 revealed MOG-IgG findings at the individual time points of longitudinal tests according to the relapse and remission status. After exclusion of post-steroid pulse therapy samples, all of the MOG-IgG serological statuses at clinical relapse were positive.
MOG-IgG findings at the individual time points of longitudinal tests according to the relapse and remission status, after exclusion of poststeroid pulse samples (p-value=0.0022 by Fisher’s exact test, follow-up samples for 10 patients were available)
Four of seven samples from the time of relapse and poststeroid pulse therapy converted MOG-IgG negative (figure 3F,G,I). Two patients positive for MOG-IgG underwent plasmapheresis, and after plasmapheresis, the serological status level of MOG-IgG decreased (figure 3G,H). Three of six persistent patients positive for MOG-IgG were treated with azathioprine (figure 3C,D,F), while all of the four patients that became seronegative were treated with rituximab or had history of treatment with mitoxantrone (figure 3G–J).
Discussion
The patients positive for MOG-IgG of our large unselected cohort with CNS inflammatory diseases had diverse clinical phenotypes and courses. In this cohort, MOG-IgG was detected in 4.4% (22/505) of patients and in 4.1% (10/243) of patients with NMOSD who fulfilled the IPND criteria. Seroprevalence of MOG-IgG was comparable with that of one previous study based on an unselected cohort with CNS inflammatory diseases (17/270, 6.3%),25 but was slightly lower than those of previous studies based on the selected NMOSD cohorts, which varied from 5.6% to 38%.7–13 However, the prevalence of NMOSD in patients with MOG-IgG could not be directly compared because the previous study was performed before introduction of the IPND criteria.
Not surprisingly, the NMOSD clinical phenotype according to the IPND criteria was most commonly observed in patients with MOG-IgG (10/22, 45.5%), comparable with a recent study (16/50, 32%).14 Double positivity for both MOG-IgG and AQP4-IgG was rarely observed in patients with NMOSD (0.4%), consistent with previous studies using CBAs (1%–2.9% double positivity in patients with NMOSD).7 12 21 These patients have typical clinical manifestations of NMOSD and the added impact of MOG antibodies in this context is unclear. The double-positive patient in our cohort had the highest EDSS of any of the patients positive for MOG, but this may simply be related to the AQP4-IgG-mediated damage. Longitudinal studies on double-positive patients may help clarify their temporal association. Previous studies have demonstrated that AQP4-IgG autoimmunity was associated with astrocyte damage, while MOG-IgG autoimmunity was usually associated with myelin injury; therefore, using the term NMOSD in patients with MOG-IgG is a matter of debate.34 35
Notably, based on a longitudinal analysis over 2 years, we observed that patients did not experience a clinical relapse after conversion to seronegativity under maintenance immunotherapy, but a relapsing clinical course was observed in a subset of patients with a persistent MOG-IgG-positive status. After exclusion of acute treatment effect, MOG-IgG was positive in all serum samples that were obtained during relapses, consistent with previous studies.9 13 18 19 These findings give us a good reason to follow-up patients positive for MOG-IgG under immunotherapy, but the one patient who returns to seropositive status does encourage caution. In previous studies,16 18–20 23 negative serological conversion of MOG-IgG was observed in patients with a benign clinical course that included monophasic features. However, other studies report that patients with negative serological conversion of MOG-IgG do experience clinical relapse.12 24 Additionally, a persistent MOG-IgG-positive status has been observed in patients with relapsing clinical courses,9 10 17 20 but not always.12 20 24 These contradictory results may be explained by study differences in sample size, follow-up duration and analysis of the treatment effect.
Interestingly, changes of MOG-IgG status tended to be influenced by acute or maintenance therapeutic regimen. A MOG-IgG-negative status was observed in four of seven serum samples at the relapse status after steroid pulse therapy, and MOG-IgG CBA scores decreased after plasmapheresis. All four patients that became MOG-IgG seronegative had a history of potent immunotherapy that included rituximab or mitoxantrone. However, two patients with a persistent MOG-IgG-positive status despite rituximab therapy experienced clinical relapses. Similar findings were observed in several previous studies,14 18 19 but future studies with larger cohorts and an individual therapeutic regimen are necessary to clarify the therapeutic implication of MOG-IgG status in clinical practice.
The presence of MOG-IgG in adult patients with MS has been debated.4 6 7 11 12 15 16 25 30 This is largely due to assay methodology. Most recent studies, where testing against native protein in cell-based diagnostic systems was utilised, demonstrated very low or no reactivity to MOG-IgG in adult patients with MS.7 11 12 15 16 25 30 However, a recent study showed that MOG-IgG could be detected in a subgroup of adult patients with MS (1% in a unselected MS cohort and 5% in a selected MS cohort with concomitant initial spinal cord and brainstem involvement). All patients in this subgroup showed a typical relapsing-remitting clinical course and two of these patients stabilised with natalizumab therapy.24 In the present study, one adult patient (0.8% of adult MS patients) showed the clinical and MRI features consistent with relapsing MS, and CSF-restricted oligoclonal bands. She responded well to disease-modifying therapy for MS, but after discontinuation of therapy, she experienced a series of relapses, which subsided after re-administration of disease-modifying therapy. She appeared to become MOG positive during this treatment break after three relapses and reverted to seronegative between 6 and 15 months later. However, the only available samples at earlier time points, which were negative, were poststeroid pulse-therapy.
Retrospective design of the current study entails several methodological limitations including inconsistent and incomplete sampling. Additionally, only 2% of the samples (11/505) were obtained at clinical onset; therefore the clinical implications of MOG-IgG status during an early phase of disease remains unknown. Lastly, the inclusion of patients from a single referral centre and patients with available serum samples resulted in an unintentional selection bias, including the preferred enrollment of patients with relapsing clinical courses and/or high disease activity. For the same reason, no paediatric ADEM or monophasic isolated optic neuritis patient with MOG-IgG could be enrolled. Further prospective multicentre studies that include both adult and paediatric patients and regular assessment for MOG-IgG status with longer follow-up from an early stage of disease are warranted.
In conclusion, we confirmed that the clinical phenotype of NMOSD was most commonly observed in patients positive for MOG-IgG from a large unselected cohort with CNS inflammatory diseases. Measurements during long-term follow-up suggested that persistent positivity of MOG-IgG is associated with clinical relapses.
References
Footnotes
PW and HJK are joint senior authors
Contributors HJK and PW had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: JWH, SHK, HJK and PW. Acquisition, analysis or interpretation of data: all authors. Drafting of manuscript: JWH, HJK and PW. Critical revision of manuscript for important intellectual content: all authors. Study supervision: HJK and PW.
Competing interests JWH, MRW, SHK, IHJ, BSK, GYK, YSK report no disclosures. SRI is supported by the Wellcome Trust, British Medical Association Research grant, Vera Down grant and Epilepsy Research UK. PW has received honoraria from Biogen Idec Japan, Euroimmun AG, Germany and Mereo Biopharma, UK; SRI and PW are named coinventors and receive royalties for assays for the detection of antibodies. HJK has lectured, consulted and received honoraria from Bayer Schering Pharma, Biogen, Genzyme, HanAll BioPharma, MedImmune, Merck Serono, Novartis, Teva-Handok and UCB; received a grant from the Ministry of Science, ICT & Future Planning; accepted research funding from Genzyme, Kael-GemVax, Merck Serono, Teva-Handok and UCB; serves on a steering committee for MedImmune; is a coeditor for the Multiple Sclerosis Journal—Experimental, Translational and Clinical, and an associated editor for the Journal of Clinical Neurology.
Patient consent Obtained.
Ethics approval The Institutional Review Board of NCC approved the present study.
Provenance and peer review Not commissioned; externally peer reviewed.
Correction notice Since this article was first published online an update has been made to the author name Byeong-su Kong. This author name has been updated to Byungsoo Kong.
Linked Articles
- Editorial commentary