Article Text

Abstract
Objective To analyse clinicoepidemiological features of neuromyelitis optica in a large cohort and to compare the differences between onset age, gender and clinical phenotypes.
Methods Antiaquaporin-4 antibody (AQP4-ab) levels were tested in 2366 serum samples of patients diagnosed as having central nervous system inflammatory demyelinating disorders by their referring physicians. AQP4-ab was measured by indirect immunofluorescence staining using human AQP4-transfected HEK 293 cells. A blinded analysis was performed and was combined with clinical information.
Results A total of 583 patients (91.4% women) were AQP4-ab-positive. The average onset age was 42.9±15.9 years. According to MRI studies, spinal-cord lesions were detected in 85.3% of the patients, longitudinally extensive transverse myelitis in 72.7% and cerebral lesions in 51.1%. Unilateral or bilateral blindness was observed in 16.2% of patients, 19.8% were associated with Sjögren syndrome, and 13.6% were associated with thyroid diseases. Myelin basic protein was detected in the cerebrospinal fluid of 57.5% patients. In addition, men presented with an older onset age, a greater number of brainstem MRI lesions and positive myelin basic protein in the cerebrospinal fluid. All child-onset patients (<15 years, n=9) presented with optic neuritis as the first symptom, while older-onset patients presented with myelitis. Twenty patients initially developed limited brain lesions, and seven of these patients did not develop optic or spinal lesions during the 1–5-year follow-up period.
Conclusions The clinical characteristics of AQP4-ab-positive patients were similar. However, optic neuritis was more common in paediatric patients, while myelitis was more common in older patients. A small number of patients exhibited only cerebral, brainstem, or cerebellar lesions during the initial several years and lower Extended Disability Status Scale scores.
- Neuroimmunology
- multiple sclerosis
- parkinson's disease
- CSF
- infectious diseases
- steroids
Statistics from Altmetric.com
Neuromyelitis optica (NMO) is an inflammatory demyelinating disease of the central nervous system that primarily affects the optic nerve and spinal cord with highly specific autoantibody NMO-IgG levels seen in the serum of NMO patients.1 The target antigen of NMO-IgG has been identified as aquaporin-4 (AQP4), a water-channel protein predominantly expressed in the central nervous system.1 2 NMO-IgG expression is restricted not only to typical NMO but also to NMO spectrum disorders (NMOsd) as well that include the following clinical features3: (1) limited disease forms, such as optic neuritis (ON) or longitudinally extensive transverse myelitis (LETM); (2) atypical brain lesions with characteristics typical for NMO and positive NMO-IgG; and (3) ON or LETM associated with a systemic autoimmune disease.
An increasing number of studies have reported the clinicoepidemiological features of NMO,4–9 although the patient number in each study has been insufficient. Therefore, the population prevalence of NMOsd has not yet been firmly established. The present study analysed clinical and epidemiological features in a large group of Japanese patients with NMO. Anti-AQP4 antibody (AQP4-ab)-positive or AQP4-ab-negative patients who exhibited characteristic NMO clinical features were compared and analysed under identical conditions at a single institute using an antibody assay system.
Patients and methods
Patients
A total of 2366 serum samples were sent to our laboratory from other Japanese institutions during 2006–2009 for AQP4-ab testing. All patients were diagnosed as having inflammatory demyelinating disorders of the central nervous system by their referring physicians by MRI findings and laboratory tests. Clinical information was obtained under informed consent from patients by their referring physicians. According to the clinical records, the patients exhibited features such as ON, myelitis, and cerebral or brainstem lesions, although no inclusion or exclusion criteria were set for the AQP4-ab test. The present study provided the definition of ‘severe impairment’ of vision when patient eyesight was worse than 20/200. To avoid quantification of false-negative cases, AQP4-ab was considered negative when the sample was collected during the acute phase of the disease or within a month of exacerbation, and no plasmapheresis or immunosuppressive therapy was performed prior to the sampling. Analysis included gender, age at onset, involved lesions of the first attack and relapses, number of relapses, Expanded Disability Status Scale (EDSS) score at the time of the AQP4-ab test, vision outcome, detected lesions on MRI scans, history of systemic disease, serum auto-antibodies as antinuclear antibody, anti-Sjögren syndrome A antibody (SS-A) or anti-Sjögren syndrome B antibody (SS-B) and others, myelin basic protein (MBP) expression, and oligoclonal IgG bands (OCB) in cerebrospinal fluid (CSF). OCB was measured by isoelectric focusing (Mitsubishi Chemical Medience, Tokyo, Japan) or agarose gel electrophoresis (SRL, Tokyo, Japan), while MBP was measured by enzyme immunoassay (Mitsubishi Chemical Medience) or ELISA. According to these results, the patients were assigned to four subgroups: NMO type (patients with both ON and LETM), ON type (presenting with only relapsing ON more than three times), spinal type (presenting with isolated recurrent myelitis more than three times with LETM on MRI) and brain-dominant type (presenting cerebral, brainstem or cerebellar lesions, but not ON or myelitis during relapsing clinical course). AQP4-ab-negative patients who presented with both LETM and severe ON (seronegative NMO) were also analysed and compared with the AQP4-ab-positive patients with LETM and/or severe ON (seropositive NMO).
AQP4-ab testing methods
AQP4-ab was measured by indirect immunofluorescence staining using human AQP4-transfected human embryonic kidney (HEK) 293 cells as previously reported.10 In brief, cDNA encoding the full-length human AQP4 was ligated to an expression vector. The vector was then transfected into HEK 293 cells, which were grown in a poly-l-lysine-coated chamber slide using Lipofectamine reagent (Invitrogen, Eugene, Oregon) according to the manufacturer's instructions. At 24 h post-transfection, HEK-293 cells were fixed in 4% paraformaldehyde/0.1 M phosphate-buffered saline (PBS, pH 7.4) for 20 min. Non-specific binding was blocked with 10% goat serum/PBS, and the cells were incubated with patient sera (1:100; if staining was negative, a 1:10 dilution was also tested) followed by fluorescein isothiocyanate-conjugated antihuman immunoglobulin G (IgG). SlowFade Gold antifade reagent (Molecular Probes, Eugene, Oregon) was applied to the slides, and staining was observed via fluorescence microscopy. To ensure antibody assay specificity, AQP4-ab was measured in 10 healthy subjects, 40 non-neuroinflammatory disease patients (cerebral infarction, spinocerebellar degeneration, amyotrophic lateral sclerosis, etc) and 30 typical multiple sclerosis (MS) patients who exhibited a relapsing-remitting clinical course with typical MRI findings (ovoid lesions and Dawson's finger). All subjects were negative for AQP4-ab, suggesting that the test was highly specific. In the present study, antibody tests were performed blindly with no clinical information being given to the examiner.
Statistical methodology
A statistical analysis of onset age, EDSS, disease duration and annual relapse rate was performed using the multiple variation of the Tukey honestly significant difference test between the four phenotype groups, the Mann–Whitney U test between the two seropositive/seronegative groups and the Kruskal–Wallis H test to compare the five subgroups according to age of onset. Statistical differences were tested for significance using the χ2 test in each subgroup. Between the five subgroups of onset ages, statistical significances were confirmed using the χ2 test, while statistical differences were tested using the Cochran–Armitage trend test.
Results
A total of 583 patients (502 women and 47 men; detailed clinical data were unavailable for the other 34 patients) were AQP4-ab-positive. The average age at onset was the early 40s, the average EDSS score was 5.6, and the average annual relapse rate was 1.4. First-attack lesions in the optic nerve and spinal cord occurred in 49.0% and 46.4% of the patients, respectively. According to MRI results, spinal-cord lesions were present in 85.3% of patients, including LETM in 72.7% and brain lesions in 51.1%. Unilateral or bilateral blindness was detected in 16.2% of patients, and severe visual impairment was seen in 28.6%. Sjögren syndrome (SjS) was associated with 19.8% of patients, and thyroid diseases including chronic thyroiditis, Graves' disease or benign thyroid tumours were seen in 13.6%. CSF OCB and MBP were positive in 12.0% and 57.5% of the patients, respectively. When genders were compared, the average onset age was greater in men (49.9 in men vs 42.2 in women). In addition, the number of brainstem lesions (MRI) (60.4%) and CSF MBP (86.6%) levels was significantly greater in men.
A total of 337 of the AQP4-ab-positive patients met McDonald's diagnostic criteria for MS 2005.11 Among these 337 patients, 251 developed LETM throughout the clinical course, 42 had spinal cord lesions but no LETM, and 23 showed no spinal cord lesions (sufficient data on MRI were not available in 21 patients).
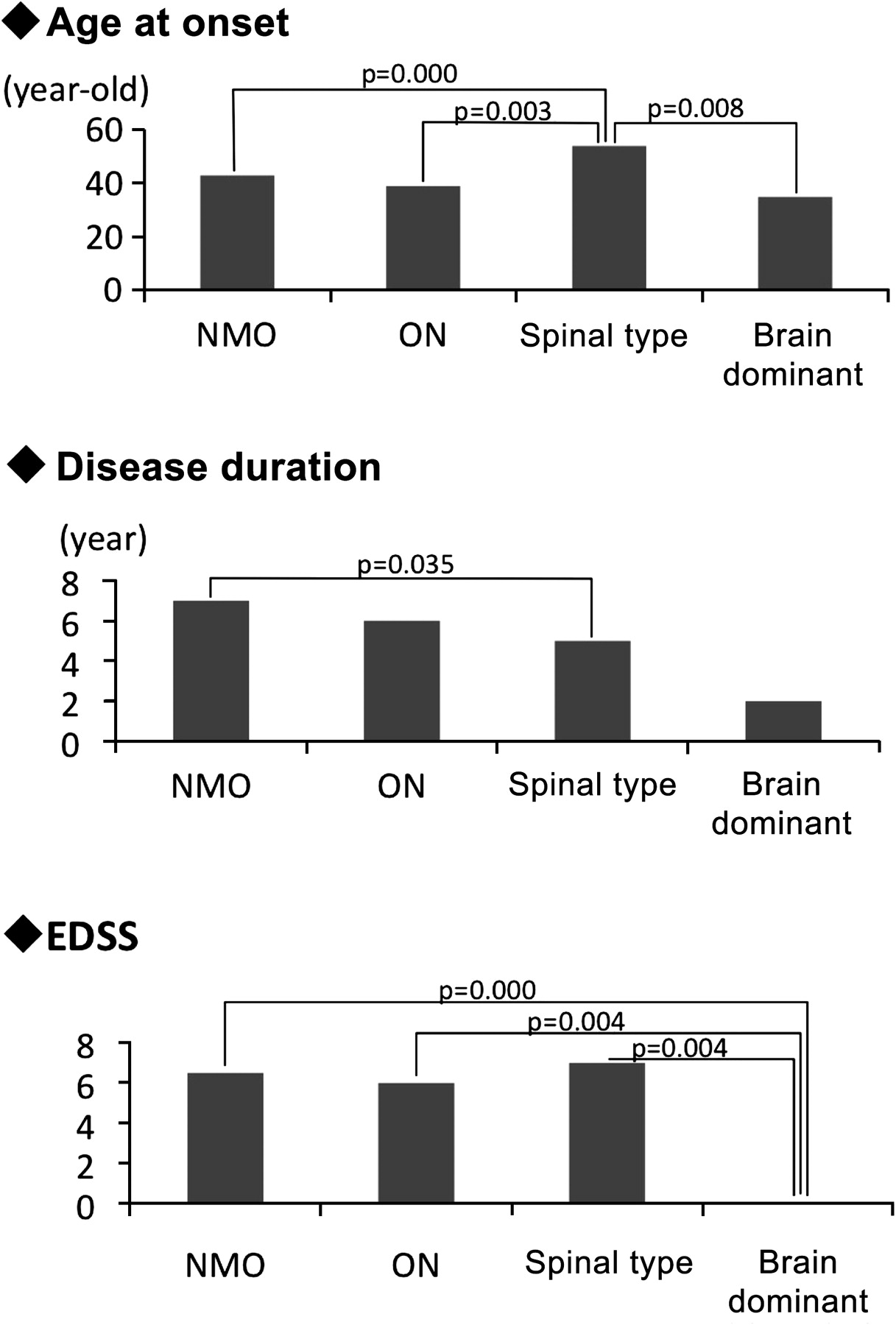
Table 1 and figure 1 show the comparison between the four clinical phenotypes: NMO type, ON type, spinal type and brain-dominant type. Gender-based differences were not observed among the four groups. Onset age was statistically higher in the spinal type than in the other three phenotypes. Disease duration was shorter in the spinal type in this study. The annual relapse rate showed no differences. The EDSS score was low in the brain-dominant type. SjS more frequently afflicted ON-type patients, while thyroid disease was more often observed in the brain-dominant type. CSF OCB positivity was greater in the brain-dominant type.
Comparison of clinical features of four phenotypes with aquaporin-4 antibody-positive neuromyelitis optica patients

Comparison of the clinical features of aquaporin-4 antibody-positive patients among the four phenotypes. The median onset age was statistically higher in the spinal type than in the three other phenotypes. The Extended Disability Status Scale (EDSS) was higher in the neuromyelitis optica (NMO), optic neuritis (ON) and spinal types than in the brain-dominant type, reflecting the severity of optic neuritis or myelitis.
In addition, AQP4-ab-positive patients were compared according to onset age (figure 2). Child-onset patients, comprising nine patients <15 years, were women and displayed ON as the initial symptom. A high female-to-male ratio and ON onset were more predominant in the younger subgroups. In contrast, the ratio of spinal cord lesions as the initial symptom was greater in the older subgroups.
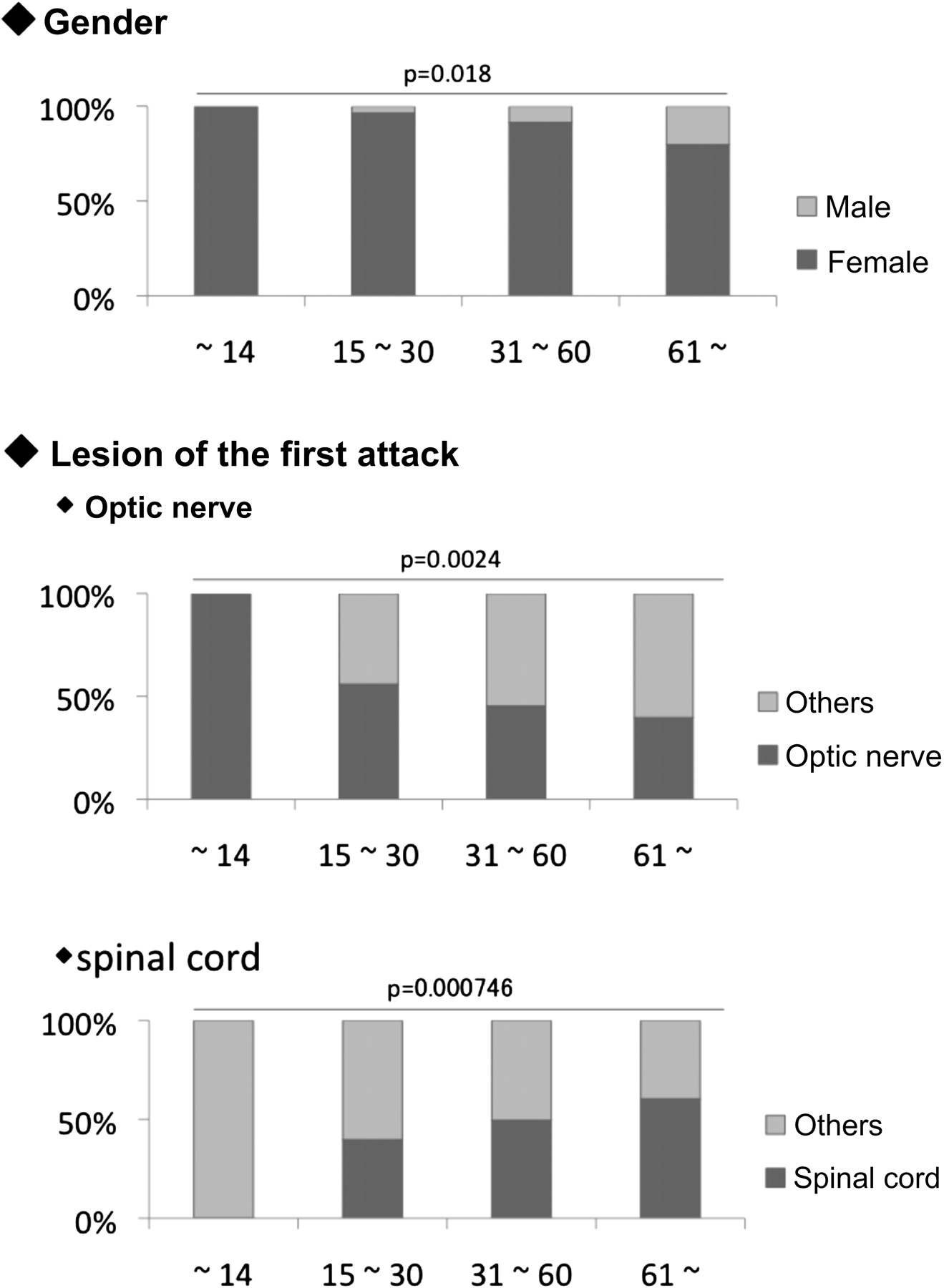
{kind=link}
{kind=link}
Comparison of aquaporin-4 antibody-positive patients of different age groups. A total of nine paediatric-onset patients were included in the first group (onset age <15 years), 111 in the second (onset age 15–30 years), 291 in the third (onset age 31–60 years) and 60 in the fourth (onset age ≥61 years). All nine paediatric-onset patients were women and presented with optic neuritis as an initial symptom. Myelitis was the most common initial symptom during the later age onset. Statistical differences were tested using the Cochran–Armitage trend test.
Those who showed bilateral ON at onset were counted as two out of six in the first group (onset age <15 years), eight out of 44 in the second group (15–30 years), 24 out of 112 in the third group (31–60 years) and six out of 20 in the fourth group (≥61 years).
A total of 480 patients were AQP4-ab-negative according to the blood sampling during the acute phases of disease, none of whom had been given immunosuppressive therapy or plasmapheresis prior to the sampling. Among them, some patients met the Wingerchuk NMO criteria12 with severe ON and LETM, so we designated these patients as seronegative NMO and compared the clinical features with those of seropositive NMO patients. In total, 382 patients were seropositive NMO, and 19 were seronegative NMO. Results revealed a female-to-male patient ratio of 90.0% and 73.6% in the seropositive and seronegative NMO patients, respectively, with an onset age of 43.4±15.7 years (median 45; range 6–86 years) versus 34.8±15.3 years of age (median 32.5; range 15–70 years). In addition, the annual relapse rate was 1.3±1.0 versus 1.4±1.2, with no significant difference between the groups. In comparison, 52.6% of seropositive NMO patients and 11.1% of seronegative patients were antinuclear antibody-positive. There were no statistically significant differences between the numbers of patients with SjS or thyroid diseases, positive SS-A and/or SS-B, and OCB or MBP in the CSF samples between groups (table 2).
Comparison between seropositive and seronegative neuromyelitis optica
The present study analysed 57 AQP4-ab-positive patients presenting with cerebral, brainstem and/or cerebellar lesions as initial involvement that were not typical NMO lesions. Of these 57, 52 patients presented with a relapsing-remitting clinical course. Cerebral lesion onset was observed in 20 patients, brainstem lesions in 22, cerebellar lesions in two, cerebral/brainstem lesions in six, cerebral/cerebellar lesions in one and cerebral/brainstem/cerebellar lesions in one. At the time of this study, eight AQP4-ab-positive patients had not developed ON or myelitis during the 1–5-year relapsing-remitting clinical disease course (table 3).
Profile of aquaporin-4 antibody-positive patients in relapsing-remitting clinical course without optic neuritis or myelitis
Discussion
The present study investigated clinical data from 583 AQP4-ab-positive Japanese patients, and the results revealed an extreme predominance in women, with a higher onset age, LETM, and severe vision loss as the main clinical features, as well as a high prevalence of association with SjS or thyroid diseases.
Comparing clinical phenotypes, we found 31 AQP4-ab-positive patients with brain-dominant type presented with markedly lower EDSS scores than the others. This might suggest that the brain-dominant type has a better clinical prognosis, although this finding was obtained from a small group of patients with shorter disease duration. As such, further observations are needed to draw any conclusions.
The initial symptoms of AQP4-ab-positive patients differed according to age of onset. A total of nine patients with an onset age of <15 years exhibited ON and severe visual disturbances as initial symptoms. Patients with a higher age of onset tended to exhibit myelitis as the initial symptom. Previous reports of NMOsd discussed the differences between ethnicities or genders, but onset symptom differences between the younger and older ages were not fully understood.13 In the present study, eight of nine patients with an onset age <15 years exhibited a relapsing course, while three displayed repeated ON, suggesting that the underlying pathological mechanisms might differ between the paediatric- and adult-onset groups. However, there were study limitations between the paediatric- and adult-onset patients because of the small number of paediatric patients.
A total of 480 AQP4-ab-negative patients were identified in this study. Because there were no exclusion criteria for the AQP4-ab test, this cohort could be heterogeneous. However, 19 of the AQP4-ab-negative patients presented with LETM and severe vision loss, which was typical for the NMOsd group. Nakashima et al reported no blindness in Japanese NMO-IgG-negative patients with ON and myelitis (OSMS)14, but two seronegative NMO patients were blind in at least one eye in the present study.
In general, seropositive NMO patients exhibited different epidemiological features from the seronegative NMO patients. For instance, seronegative NMO patients exhibited less female predominance or a younger onset age than the seropositive NMO group, a finding more similar to conventional MS. However, further testing revealed that some seropositive NMO patients became antibody-negative following intensive treatment or a longstanding or chronic inactive stage. In addition, it is possible that NMO patients presented with very low antibody titres that were under the antibody-detection threshold of a conventional assay system. These results suggested that the seronegative NMO could include ‘inactive or low AQP4-ab titre’ NMO and non-NMO patients.
Furthermore, it is important to note that some AQP4-ab-positive patients exhibited symptoms other than ON or myelitis. A total of 57 patients initially presented with lesions in the cerebrum, brainstem, cerebellum or a combination. Of these 57 patients, 52 suffered from a relapsing disease course, while eight did not exhibit ON or myelitis at the time of the study (disease duration 1–5 years). Brainstem lesions have been reported to extend to severe cervical myelitis3 15, but 31 patients in this study presented with limited brainstem lesions as an initial attack (no spinal lesions). Pittock et al reported that brain MRI abnormalities were seen in 60% of NMO patients, although most were non-specific, and the symptoms of brain involvement were subtle. In the present study, 20 patients exhibited limited cerebral lesions as initial symptoms. Four of them started with cerebellar symptoms, while three showed abnormalities in the cerebellum on MRI scans. However, most patients (47 of 55) who started with atypical lesions for NMO initially developed relapsing ON or myelitis, including LETM, during the following clinical course of disease. Appropriate disease preventions are necessary during the early stages of NMO. For that purpose, the AQP4-ab test is crucial for patients exhibiting atypical features of NMO but having common pathophysiological mechanisms.
Acknowledgments
The authors thank S Honda of the Department of Public Health, Nagasaki University Graduate School of Biological Sciences, for providing advice regarding the statistical analysis methods.
References
Footnotes
Funding This study was supported in part by Health and Labor Sciences Research Grants for Research on Psychiatry and Neurological Diseases and Mental Health (KT). Grant no H-021.
Competing interests None.
Patient consent Obtained.
Ethics approval Ethics approval was provided by the ethical committee of Kanazawa Medical University.
Provenance and peer review Not commissioned; externally peer reviewed.