Article Text

Abstract
Tremor is a common neurological condition in clinical practice; yet, few syndromes are widely recognised and discussed in the literature. As a result, there is an overdiagnosis of well-known causes, such as essential tremor. Many important unusual syndromes should be considered in the differential diagnosis of patients with tremor. The objective of this review is to provide broad clinical information to aid in the recognition and treatment of various unusual tremor syndromes in the adult and paediatric populations. The review comprised of a comprehensive online search using PubMed, Ovid database and Google Scholar to identify the available literature for each unusual tremor syndrome. The review includes fragile X-associated tremor/ataxia syndrome, spinocerebellar ataxia type 12, tremors caused by autosomal recessive cerebellar ataxias, myorhythmia, isolated tongue tremor, Wilson's disease, slow orthostatic tremor, peripheral trauma-induced tremor, tardive tremor and rabbit syndrome, paroxysmal tremors (hereditary chin tremor, bilateral high-frequency synchronous discharges, head tremor, limb-shaking transient ischaemic attack), bobble-head doll syndrome, spasmus nutans and shuddering attacks. Rare tremors generally present with an action tremor and a variable combination of postural and kinetic components with resting tremors less frequently seen. The phenomenology of myorhythmia is still vague and a clinical definition is proposed. The recognition of these entities should facilitate the correct diagnosis and guide the physician to a prompt intervention.
- TREMOR
- MYOCLONUS
- MOVEMENT DISORDERS
- CLINICAL NEUROLOGY
- CEREBELLAR DEGENERATION
Statistics from Altmetric.com
Introduction
Tremor is defined as a rhythmic involuntary movement of one or several regions of the body.1 Tremor represents the most common neurological sign, as everyone has a ‘physiological’ tremor, which can only be measured with instrumental tools.
In spite of such high prevalence, few tremor syndromes are commonly recognised and discussed in literature (tables 1 and 2 display the principal features of these common tremors), thus contributing to an overdiagnosis of conditions such as essential tremor (ET), due to an ‘epidemiological bias’.2 This review will address the unusual syndromes that should receive the neurologist's attention in the differential diagnosis of patients with tremor, in light of the well-known rule: ‘know in order to recognise’ (for the search criteria, see online supplementary material).
Definitions and clinical features of common tremors
Drugs and toxins known to cause tremor; newborns might also present with tremor if the mother is taking these drugs (direct effect or withdrawal)
Supplemental material
Unusual tremor syndromes
Genetic cerebellar disorders
Intention tremor is commonly seen in patients with acquired cerebellar diseases (table 1), but it may also be a prominent feature of several genetic conditions. Tremor may occur in different spinocerebellar ataxias (SCAs), such as a resting tremor as part of a parkinsonism phenotype in SCA2 or 3 and palatal tremor in SCA20 or, more rarely, SCA7.3 Interestingly, in fragile X-associated tremor/ataxia syndrome (FXTAS) and SCA12, a tremor is often the main presenting feature with ataxia developing later on.4 ,5 Other cerebellar conditions may also have a tremor as either a unique feature observed (eg, ataxia-telangectasia or Friedreich's ataxia) or its presence may assist in the differential diagnosis between disorders (other autosomal recessive ataxias).6
Fragile X-associated tremor/ataxia syndrome
FXTAS is a progressive, late-onset neurodegenerative disorder associated with the FRM1 gene premutation (55–200 CGG repeat expansion).7 The phenotype presents typically in males with onset in the sixth decade and includes additional cognitive deficits (executive and memory dysfunction as well as global cognitive deficits), variable degrees of peripheral neuropathy, and occasionally mild parkinsonism. Psychiatric problems, such as depression or anxiety, are quite common. FXTAS encompasses two main clinical features: cerebellar ataxia and/or intention tremor where the tremor is usually the first sign with ataxia following 5–7 years later.5 ,8 Notably, up to 20% of patients with FXTAS have no clinically observable tremor. The tremor is heterogeneous but three tremor subtypes have been described:5
Cerebellar intention tremor (3–5 Hz),
ET-like,
Parkinsonian tremor.
In most cases, the presentation may be confused with ET in the early stages, especially if the tremor is mild, only tandem gait is affected, and the cognitive deficit is mild.5 In contrast, a pure resting tremor is rarely observed (6–10%).
FXTAS is associated with specific MRI findings which correlate with pathological findings.5 Increased T2/FLAIR signal hyperintensity are present in both the middle cerebral peduncle (MCP sign) and the splenium of the corpus callosum in 64% and 68% of patients, respectively (figure 1A).5 Females can be also affected, but they usually have a more benign phenotype, mainly characterised by parkinsonism and absent or mild MRI findings, usually involving the corpus callosum rather than the middle cerebellar peduncle.5
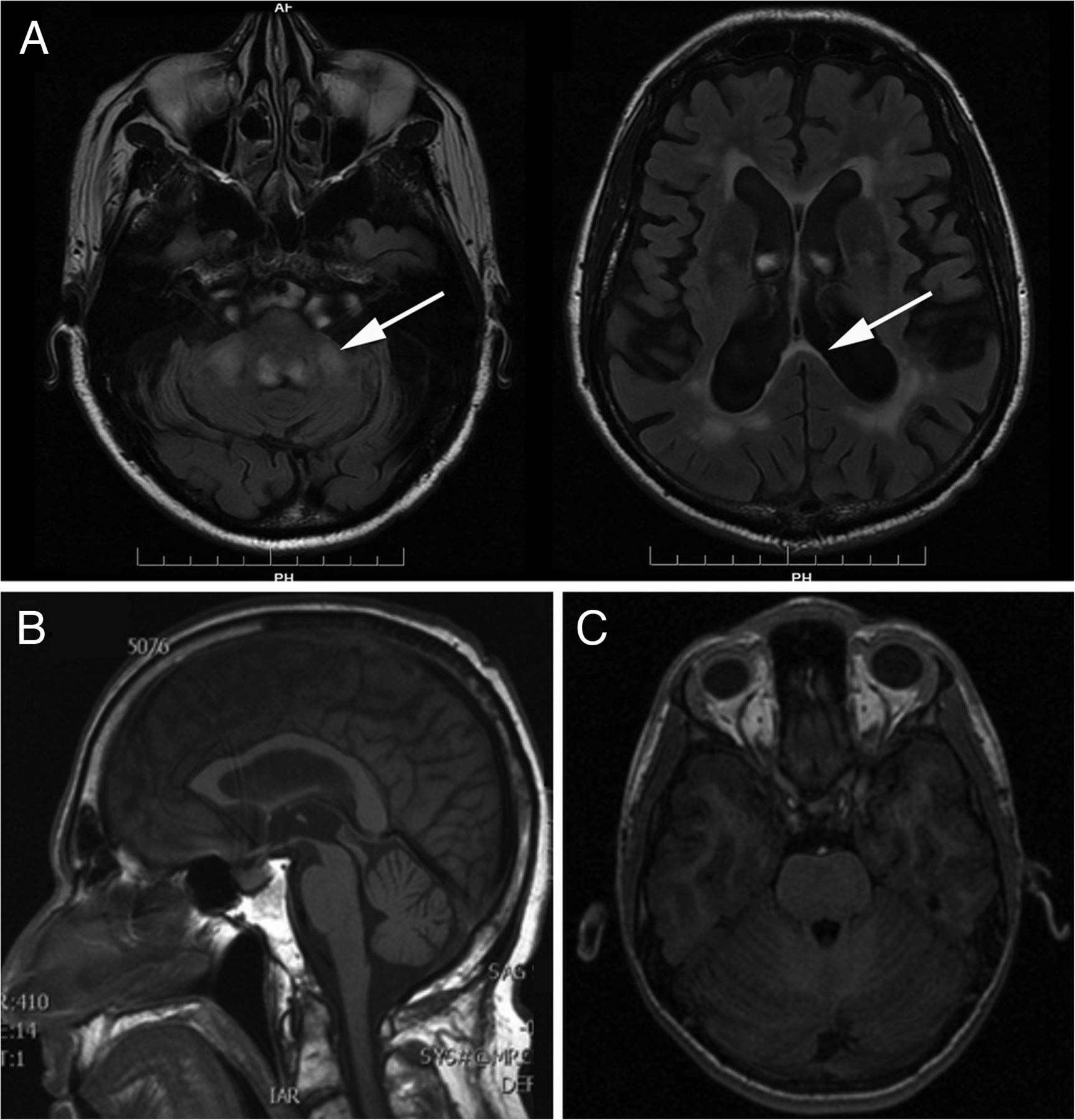
MRIs of patients with fragile X-associated tremor/ataxia syndrome (FXTAS), spinocerebellar ataxia 12 (SCA12) and ataxia-telangiectasia. (A) FXTAS is associated with an increased T2 signal intensity of the middle cerebellar peduncle and/or of the corpus callosum splenium. (B) Brain MRI of a patient with SCA12 showing widespread cerebral and cerebellar atrophy without specific focal signal abnormalities. (C) Brain MRI of a patient with ataxia-telangiectasia; no abnormalities are detectable in the posterior fossa.
It has been suggested that genetic testing for FXTAS premutations should be considered in patients presenting with an intention tremor of unknown cause in individuals over the age of 50 with concomitant parkinsonism or cognitive decline.8
The treatment of FXTAS is usually disappointing. The cerebellar or ‘essential-like’ tremors might respond to medication used for ET, including primidone, benzodiazepines5 or unilateral deep brain stimulation of ventral intermediate nucleus of the thalamus, although in our experience, surgery can also worsen the underlying ataxia without improving the tremor.9 The parkinsonian tremor is usually unresponsive to L-dopa in spite of an impaired dopaminergic system, as detectable by the dopamine transporter single photon emission CT.5
SCA12
Owing to the high frequency of tremor at disease onset in patients with SCA12, Rossi et al4 suggested that clinicians might benefit from using this feature to guide the selection of genetic testing, but cautiously due to the high level of phenotype-genotype overlap and occasional unusual cases. SCA12 is an autosomal-dominant degenerative cerebellar disorder that results from a CAG repeat mutation in the supposed upstream promoter region of the PPP2R2B gene. The disease features usually begin in young adulthood and it is often associated with an action tremor (∼3 Hz) of the upper limbs (often asymmetrical) and/or head at clinical presentation. Additional neurological signs that develop include hyper-reflexia, gait ataxia, mild parkinsonism and other cerebellar signs.4 ,10 ,11 The tremor usually progresses with an increase in amplitude and distribution, involving other regions such as the jaw, protruded tongue, legs or voice. However, patients typically remain functional, retaining the ability to work into their 50s and 60s. The features at the initial clinical presentation may be confused with a diagnosis of ET due to the age of onset and similar tremor characteristics, as well as a positive family history of tremors.10 This clinical syndrome has been described in different ethnic backgrounds (such as German) with some variability,11 but it is mainly found in patients with northern Indian ethnicity and should be considered especially when they also have a family history of tremor.10 ,11 Neuroimaging typically demonstrates widespread cerebral and cerebellar atrophy without specific focal signal abnormalities on MRI (figure 1B). Pharmacological treatment with β-blockers, primidone or benzodiazepines (eg, clonazepam) may be able to reduce the tremor amplitude in some cases but it may not provide any useful symptomatic benefit.10 To our knowledge, functional neurosurgery has not been used to treat these patients.
Autosomal recessive ataxias
The presence of tremor may be helpful in the clinical differentiation between the various autosomal recessive ataxias, particularly vitamin E deficiency, late-onset Tay-Sachs disease, mitochondrial recessive ataxia syndrome, ataxia-telangiectasia (AT), ataxia with oculomotor apraxia type 1 and 2, Cayman ataxia and Marinesco-Sjögren syndrome.6 The broad clinical spectrum of Friedreich's ataxia may also include a bilateral intention tremor of the upper limbs which has been consistently, albeit rarely, reported in the literature.12 A study has reported head tremor in 21 of 46 patients (45.7%) studied who had both a typical phenotype and were homozygous for the GAA repeat expansion in the FXN gene.13 However, the rhythmic oscillations or ‘titubations’ have been hypothesised to result from hypotonia of the axial muscles due to the low frequency (<1 Hz), large amplitude and omnidirectional nature.12
A tremor was originally described in two brothers with uniquely mild forms of AT with onset in young adulthood.14 The tremor predominantly occurred at rest (5–6 Hz) but there was also a slight action and postural tremor affecting the head and trunk. A recent study in a large series of patients using three-dimensional accelerometer recordings found a postural and action tremor (3–6 Hz) in the vast majority of patients with just over half also demonstrating a tremor at rest.15 The tremors were, however, only present transiently, occurring 6–19% of the time that the patients were observed. The pathophysiology of these involuntary movements is not completely understood but it is likely that the cerebellum is involved, although MRI of these patients may also be unremarkable (figure 1C).
Myorhythmia
Myorhythmia has been recently defined as a repetitive, rhythmic, often jerky movement of slow (1–4 Hz) frequency, typically affecting the cranial and limb muscles16 although the term myorhythmia was originally coined by Herz17 in 1931. The movements of myorhythmia predominantly occur at rest and are distinguishable from a parkinsonian tremor by the lack of L-dopa responsiveness, a more irregular and slower frequency, as well as an intermittent nature. However, both parkinsonian tremor and myorhythmia may be suppressed by sleep and persist or reduce during active movements and posture. Other movements were also described co-occurring with myorhythmia, such as myoclonus, tics, choreiform or ballistic movements.
The inclusion of a wide variety of muscles and movement disorders has resulted in diagnostic confusion in the subsequent literature. For instance, some of these tremors were predominantly described in patients with dystonia and would currently be more likely referred to as dystonic tremor.16 The main source of confusion results from the differential diagnosis with Holmes’ tremor or pseudorhythmic myoclonus, as these terms have been used interchangeably. Both Holmes’ tremor and myorhythmia are characterised by a tremor ‘often not as rhythmic as other tremors’ and by constant occurrence at rest and during action.1 Palatal myoclonus is another differential diagnosis to consider that may co-occur with myorhythmia.16
When myorhythmia is discussed, the majority of authors cite a case series of 24 patients published by Masucci et al.18 The case series found that the most common aetiology involved brainstem vascular disease and cerebellar degeneration due to chronic alcoholism or nutritional deficiency, with a peak onset of 50–59 years of age. The patients displayed an asynchronous and intermittent tremor predominantly at rest but also during action or posture, with a frequency between 2 and 3 Hz, similar to the original description by Herz.17 In the majority of patients, myorhythmia involved primarily the limbs, but also the face, jaw, head and tongue without palatal involvement. Ocular myorhythmia occurred in three of the patients whereas the term ‘extremity myorhythmia’ has been used when the limbs are predominantly affected.18 Although no formal neurophysiological investigation exists, electromyography (EMG) studies in patients with myorhythmia typically show pseudorhythmic discharges of motor units lasting approximately 200–300 ms with slow interburst rates of 1–4 Hz.19 Interestingly, the duration of the discharges is longer than myoclonus but certainly shorter than that of classical tremors.
Other subtypes of myorhythmia have also been classified, often adding to the confusion. Oculomasticulatory myorhythmia (OMM) has been described as a rare neurological sign of Whipple's disease characterised by rhythmic contractions of the masticulatory muscles paired with smooth ocular convergent oscillations (∼1–3 Hz). A literature review highlighted a consistent use of OMM to describe the neurological findings in multiple cases of Whipple's disease. Interestingly, several patients were diagnosed on the basis of OMM without gastrointestinal features suggesting that this feature may be pathognomonic for Whipple's disease.20
Myorhythmia generally has a poor prognosis, especially in cases resulting from vascular disease (eg, thalamic or brainstem infarcts).18 A favourable recovery may occur with treatment of the underlying condition, such as antibiotics in Whipple's disease.20
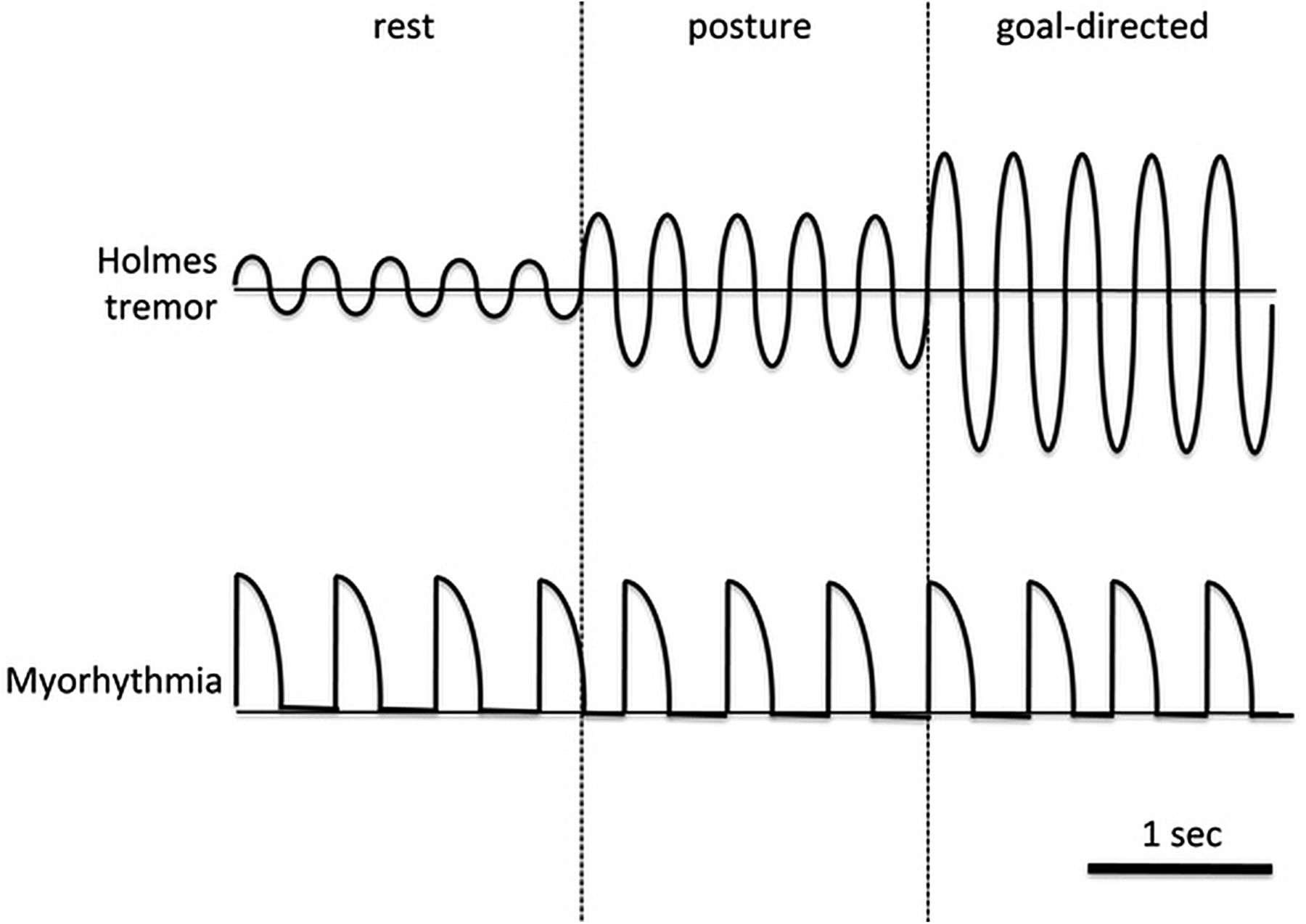
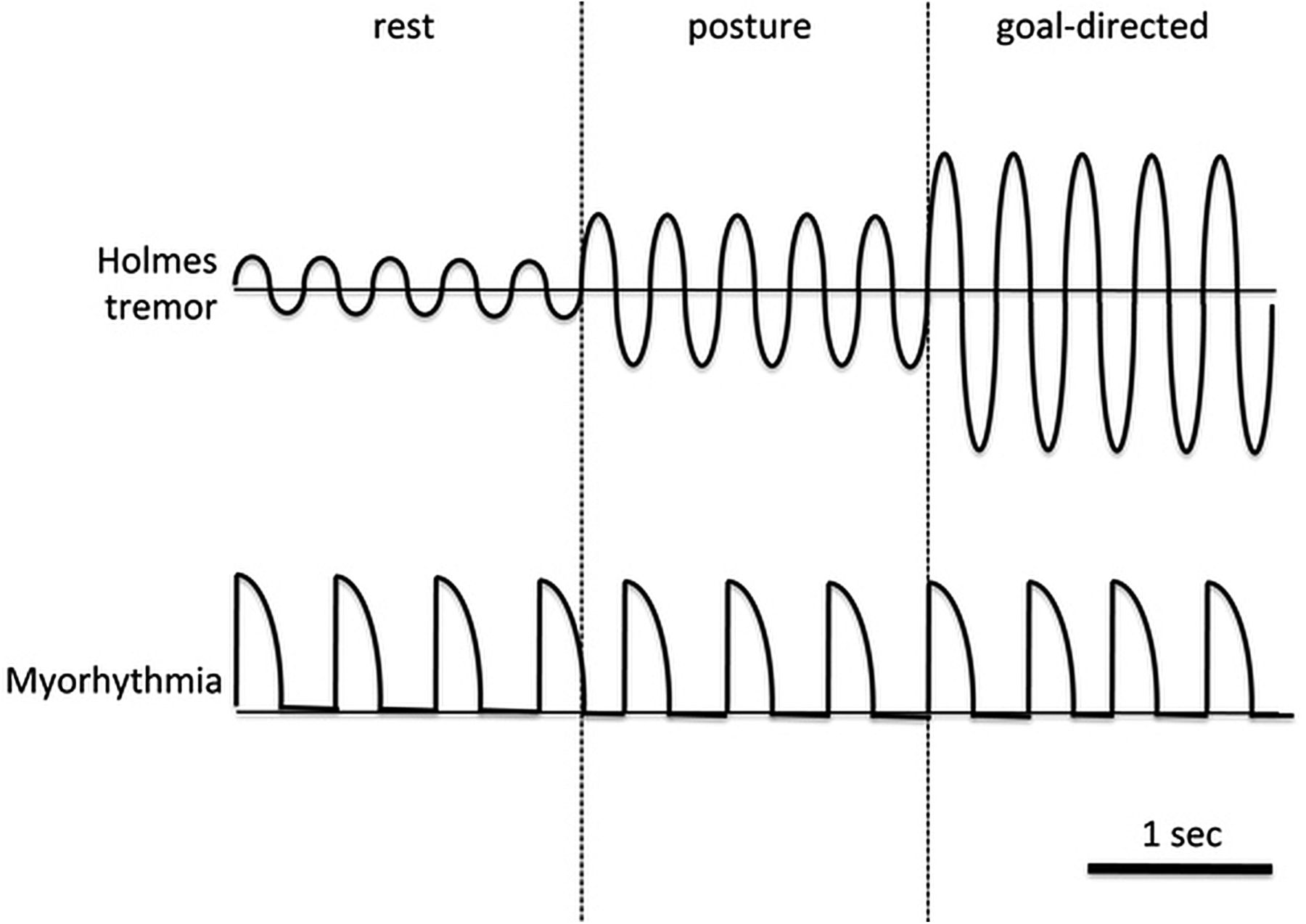
Various other conditions have been reported to be associated with myorhythmia. Table 3 summarises the existing literature on myorhythmia and also emphasises the high variability of clinical presentation of patients labelled with this condition. While conducting the literature review, available video evidence of cases reported as myorhythmia were also analysed by two authors (AF and AEL). In some cases, the original authors did not classify the movement as myorhythmia but they were included in a recent review on the topic.16 Forty-four video segments were taken into account and inter-rater agreement ranged from 79.5% (variability) to 90.9% (frequency). In all cases but one, the observed movements were either pseudorhythmic or non-rhythmic. The frequency was low (1–3 Hz) in 82.5% of the cases and variable (either in amplitude or frequency) in 85.7% of the cases. More importantly, in many cases, the clinical picture was difficult to classify due to a complex and irregular nature, but generally resembled dystonic movements or Holmes’ tremor. However, there was consistency between movements observed in several cases of Whipple's disease. Therefore, we believe that the term myorhythmia has been correctly used in the description of Whipple's disease but it has been misleading in other cases. Despite the absence of a formal neurophysiological study, we propose that myorhythmia is distinguishable on the basis of semirhythmic rapid muscle contractions (but typically more sustained than in pure myoclonus), which are very mildly and variably modulated by actions (figure 2). Overall, the irregular clinical features of myorhythmia do not conform to traditional definitions of tremor and should be considered a separate entity, such as rhythmic myoclonus.
Published cases of myorhythmia
{kind=link}
{kind=link}
Clinical comparison between Holmes’ tremor and myorhythmia. Myorhythmia is distinguishable on the basis of semirhythmic rapid muscle contractions (but typically more sustained than in pure myoclonus), which are very mildly modulated by actions. In contrast, Holmes’ tremor has a more sinusoidal behaviour and typically worsens in amplitude with voluntary movements, often to a very disabling degree. In addition, the tremor oscillates about a fixed point (the horizontal line) whereas myorhythmia usually starts and returns to the same point.
Isolated tongue tremor
Tongue tremor, accompanying other tremors and clinical features, is occasionally observed in ET, parkinsonism, or in patients with symptomatic palatal tremor. In some patients with ET or Wilson's disease, a tongue tremor may be the predominant or initial feature. The ‘trombone tremor’ of the tongue has been rarely described in neurosyphilis.21 An isolated tongue tremor is a very rare condition that has been described in brainstem or cerebellar astrocytomas, postelectrical injury or γ knife radiosurgery for acoustic schwannoma.22 ,23 An autoimmune aetiology in anti-Hu paraneoplastic conditions has been also described.24 The majority of cases are believed to be due to secondary lesions of the Guillain-Mollaret triangle. Clinically, the tremor is described as rhythmic alternating protrusion-retracting movements of 3–5 Hz, usually affecting the posterior part of the tongue with no palatal involvement. It may be observed when the tongue is protruded, at rest or during both conditions. Isolated tongue tremor has been reported to follow a self-limiting course with a duration of approximately 6 months. Several pharmacological therapies have shown subjective benefit including central anticholinergics and sodium valproate.22
Wilson's disease
Wilson's disease is a rare autosomal recessive genetic disorder of copper metabolism caused by mutations of the ATP7B gene, which results in hepatic and neurological disease. Approximately 40–50% of patients present with neurological or psychiatric symptoms, and of those, tremor is one of the main early features observed (see online supplementary material for further details).25
Slow orthostatic tremor
A ‘slow’ orthostatic tremor has been rarely reported in the literature, and it is still a poorly defined entity since the first description in a family with ET.26 Patients generally present with progressive unsteadiness and bilateral leg shaking when standing over the course of 3–6 months. Slow orthostatic tremor occurs with standing and is absent at rest or during walking. However, EMG studies of the leg or trunk muscles reveal a tremor with a frequency lower than 12 Hz (usually 4–9 Hz), which is slower than the classical orthostatic tremor (14–16 Hz).27 ,28 The tremor is distinct from orthostatic myoclonus with regular 70–120 ms bursts that are coherent with synergistic muscles. However, some asymmetry has been described between the lower limbs.29 Currently, three subtypes can be recognised:
‘Primary’ forms without overt aetiologies;30 differential diagnosis includes functional orthostatic tremor, which also presents with a frequency lower than that of classical orthostatic tremor.
‘Secondary’ forms, in the context of cerebellar and pontine lesions or autoimmune processes of the central nervous system (eg, anti-Hu and anti-Yo antibodies, especially in the context of small cell lung cancer);27 ,29 ,31 this tremor may even be the initial presenting feature in Graves’ disease.28
‘Pseudo-orthostatic tremor’, described in Parkinson's disease, Lewy body dementia and patients with SCA3: given the frequency of 4–6 Hz, the asymmetry and the response to L-dopa, it is considered a different expression of classical resting tremor.32 Another ‘pseudo’ form of the tremor is triggered by the haemodynamic changes induced by orthostatism, which represents a limb-shaking transient ischaemic attack (TIA; see below).33
The management of slow orthostatic tremor is generally with agents used in classical orthostatic tremor (eg, clonazepam) but there is usually little clinical improvement.29 In secondary cases, the tremor may significantly improve with treatment of the primary disease state, such as intravenous immunoglobulin in autoimmune cases or chemotherapy in paraneoplastic forms.27 ,31 In cases associated with Parkinson's disease, the tremor is usually responsive to L-dopa.32
Tremor induced by peripheral lesions
Tremor induced by peripheral lesions is a controversial entity, believed to be largely a functional movement disorder by many tremor experts. Nevertheless, there is some limited evidence that peripheral trauma can cause tremor (see online supplementary material for further details).
Unusual drug-induced tremors: tardive tremor and rabbit syndrome
Tardive tremor is a unique variant of a drug-induced tremor that has been associated with long-term use of agents with dopamine receptor blocking properties, such as neuroleptics and metoclopramide. It was first described by Stacy and Jankovic,34 and since then, only 10 cases have been reported in the literature.35 The cited cases include disabling tremors with coarse amplitude and frequencies ranging from 2.5 to 6 Hz, involving the upper limbs as well as the lower limbs, head, face, jaw and lips in some cases. The tremor is most severe when holding posture but also occurs at rest and during active movements. Onset of the tremor occurred between 2 and 20 years after initial drug exposure, typically developing on withdrawal of the medication, and persisting long after the removal of the offending agent. Patients usually have other coexistent tardive syndrome movements (eg, orofacial dyskinesia) with minimal or no parkinsonian features.34 When presented with a patient with a drug-induced tremor, the features that assist in distinguishing tardive from parkinsonian tremor include:
Persistence or worsening of the tremor after removal of dopamine blocking agents;
Predominantly action tremor;
Improvement with agents that antagonise the effects of dopamine, for example, tetrabenazine, increasing or reintroducing the neuroleptic.34
The risk factors for developing this tremor are not known, but many clinicians believe that patients with ET, older age and female gender have a higher risk.35
Tardive tremor should also be differentiated from the ‘rabbit syndrome’, a term first used by Villeneuve36 to describe an orofacial movement similar to that of a rabbit eating and is often associated with a popping sound. It classically occurs after long-term use (months to years) of antipsychotics antagonising dopaminergic receptors, with a prevalence of 2.3–4.4% in treated patients.37 In addition, cases unrelated to neuroleptic exposure were reported to occur with imipramine, citalopram, paroxetine, methylphenidate and phenol intoxication.38 Rabbit syndrome consists of fine rhythmic movements (5 Hz) at rest involving only the vertical axis of the oral, perinasal and masticatory muscles. It might be considered a variant of parkinsonian tremor affecting the face and should not be confused with tardive dyskinesias. Unlike tardive tremor and tardive dyskinesia, rabbit syndrome usually shows improvement with anticholinergic agents. Another, possibly better, strategy would be to switch the current antipsychotic to either quetiapine or clozapine, the latter being preferred given its anticholinergic activity.38
Paroxysmal tremors
The following tremors are characterised by their episodic nature. Spasmus nutans and shuddering attacks are other paroxysmal tremors but they will be described in the following section.
Hereditary chin tremor
Hereditary chin tremor (HCT; ‘hereditary quivering of the chin’, ‘familial geniospasm’, ‘hereditary essential chin myoclonus’) is a rare condition characterised by episodes of involuntary oscillatory rhythmic movements of the chin muscles, mainly the mentalis muscle.39 HCT is an autosomal dominant condition with high penetrance, and has been linked to chromosome 9q13-q21 in one family.40 However, the condition does have genetic heterogeneity.
HCT usually occurs at birth or early childhood with symptoms peaking in early adulthood and gradually improving or even disappearing by the fifth decade.41 The recurrent episodes last from seconds to hours and may be triggered by emotion or anxiety. The amplitude is variable and the tremor frequency varies between 2 and 11 Hz. It has been argued that neurophysiological evidence support the movements as fast, jerk-like with an irregular frequency, and thus it might be better classified as myoclonus rather than an orolingual tremor.21 ,42 The movements are generally considered benign with little interference with speech, drinking or sleep, although exceptions have been described. They may cause social embarrassment, which is the typical reason for seeking medical attention. Associated conditions have also been described, such as tongue biting, myoclonus, nystagmus, nocturnal bruxism, rapid eye movement sleep behavioural disorder, otosclerosis, familiar neuropathies and Parkinson's disease.41 ,42 Pharmacological therapies, such as benzodiazepines, haloperidol, phenytoin and hydroxyzine hydrochloride, have usually shown a poor response.41 The most effective therapy is botulinum neurotoxin injections, with positive and sustained effects with repeated injections.42
Bilateral high-frequency synchronous discharges
Bilateral high-frequency synchronous discharge (BHFSD) is a new form of tremor that has been reported in three patients with posterior fossa disorders; one case occurred secondary to a haemorrhagic lesion in the midbrain and two cases were associated with sporadic olivopontocerebellar atrophy.43 ,44 BHFSD are characterised by a brief burst (<100 ms) of highly coherent tremor of 14–16 Hz that may be triggered by maintaining the upper limbs outstretched and arrested by forceful wrist flexion.44 A concomitant low-frequency postural tremor in the upper limbs is also present. Owing to the high frequency and short duration, patients complain of episodes of involuntary vibrations or contractions in their upper limbs. The episodes ranged from several times a week to up to four times per day. Interestingly, electrical stimulation over the posterior fossa was able to reset the explosive high-frequency bursts.44 No specific treatments can be recommended but clonazepam, trihexyphenidyl and gabapentin were reported to have no benefit in one case.44
Paroxysmal head tremor
An adult-onset paroxysmal head tremor has been reported to occur with a ‘no-no’ direction of movement.45 It is a 3–5 Hz tremor involving the splenius and sternocleidomastoid muscles that occurs without any sustained directional pull of the head or neck. The onset is typically in early adulthood with the tremor becoming progressively worse over time.46 The episodes last between 5 and 60 min and may occur up to several times a week. The paroxysmal head tremor has been associated with a missense mutation in the CACNA1A gene, encoding the α 1A subunit of the calcium voltage-dependent P/Q type channel.46 Point mutations of this gene result in a variety of overlapping phenotypes including benign paroxysmal torticollis of infancy, adult-onset, focal, and segmental dystonia, episodic ataxia type 2 and familial hemiplegic migraine type 1. These conditions show clinical overlap with SCA6, generally caused by CAG repeat expansions in the coding region of the gene. The head tremor may be the predominant presenting feature in hemiplegic migraine.46 A beneficial effect of acetazolamide, clonazepam or propranolol has been reported but some cases are resistant to medical therapy.45 ,46
Limb-shaking TIA
Limb-shaking TIA is another rare condition characterised by an involuntary, typically unilateral, upper limb jerking tremor caused by a TIA.47 The episodes are transient in nature but may last several minutes. Episodes occur monthly or up to several times per day and are typically triggered by orthostatic positional change, hypotension, hyperventilation, neck extension or walking.48 The pathology is usually a severe carotid stenosis (>80%) on the side contralateral to the symptoms which is believed to result in hypoperfusion of watershed areas leading to the tremor. The presentation is often misdiagnosed as a focal motor seizure but there is a lack of a jacksonian march or aura and it does not respond to antiepileptic drugs.48 There may be other symptoms more typically seen in TIAs, such as limb weakness and speech difficulties48 or other movement disorders, such as ataxia, dystonic posturing and parkinsonism.49 Myoclonic jerks might also be associated and one patient studied with EMG during the tremor episodes was even found to have a negative myoclonus.50 It is important to identify this uncommon presentation of TIA, as these patients are at high risk for stroke. The restoration of cerebral perfusion with carotid endarterectomy, as well as intracranial angioplasty and stenting, has been associated with a resolution of the limb-shaking events.48 However, there is some concern that carotid surgery is associated with significantly higher risk of postoperative haemorrhage in patients with limb-shaking TIA than in patients undergoing carotid surgery for other indications, occurring in 23% and 0.5%, respectively.51 An alternative conservative approach for the limb-shaking events is to reduce any antihypertensive medications.49
Tremor in newborns and children
Two-thirds of healthy newborns exhibit mild tremors during the first few days of life, probably as a consequence of the immaturity of the spinal inhibitory interneurons, thus causing an excessive muscle stretch reflex.52 Two important, yet rare, syndromes to identify in the differential diagnosis of abnormal head movements in newborns or children are bobble-head doll syndrome and spasmus nutans. Shuddering (or shivering) attacks are relatively more common but their pathophysiology is still not clear.
Bobble-head doll syndrome
Bobble-head doll syndrome is a complex movement disorder in childhood first described by Benton et al53 in 1966 as a to-and-fro bobbing or nodding of the head. The syndrome mostly occurs due to an expansion in the region of the third ventricle resulting from a suprasellar arachnoid or third ventricular cyst, although it is estimated that only 10–30% of supracellar cysts result in bobble-head doll syndrome.54 ,55 Other common pathologies reported include aqueductal stenosis, malfunctioning shunts or ventricular tumours.54 Bobble-head doll syndrome usually presents in children before the age of 11 and may occur as early as 4 months.55 The main presenting feature is an episodic 2–3 Hz ‘yes-yes’ tremor of the head with occasional ‘no-no’ movements. Other areas of involvement include the neck, shoulders, trunk or upper limbs.54 The tremor is usually absent during sleep and the amplitude typically increases with walking or excitement and temporarily decreases or disappears under volitional control (eg, seconds to minutes) or concentration.55 Other presenting signs include macrocephaly, hyper-reflexia, psychomotor retardation, optic nerve atrophy and occasional endocrine dysfunction.55
The long-term prognosis is generally very good with surgical decompression resulting in complete resolution of the tremor in most cases. However, there is commonly a delay in the diagnosis as the features may be misdiagnosed as a ‘neurotic disorder’ or a tic.54 It is important to recognise and treat bobble-head doll syndrome early since a prolonged time to intervention is associated with less satisfactory outcomes, including the persistence of head tremor, psychomotor retardation, visual disturbance or endocrine dysfunction.55 It is recommended that these lesions are treated in neurosurgical centres with neuroendoscopic interventions along with long-term follow-up because residual cysts or persistent hydrocephalus may occur up to several years after the initial procedure.54 ,55
Spasmus nutans
Spasmus nutans is a paroxysmal disorder described by a clinical triad of head shaking, nystagmus, and abnormal head posture or torticollis. The head tremor in spasmus nutans is reminiscent of bobble-head doll syndrome, with a slow 2–3 Hz tremor which is also absent during sleep but usually in the ‘no-no’ direction, although it may involve other planes as well (eg, ‘yes-yes’ or rotary).56 The nystagmus is horizontal and often asymmetrical, low amplitude and high frequency but may be monocular, or vertical. It is also common to have coexistent strabismus as well. The tremor and nystagmus may decrease in severity or, in contrast, be elicited if the child visually fixates on an object. The usual onset is in infancy between 4–18 months but may be up to 3 years. The head tremor and nystagmus predominate in the majority of cases (both with paroxysmal episodes) with the abnormal head positions occurring in approximately 40%.56
The diagnosis of spasmus nutans implies an idiopathic, benign disorder with a natural history of spontaneous resolution within 1–2 years after onset and requiring no specific treatment. A variety of potential risk factors have been associated with a higher incidence of spasmus nutans including low socioeconomic status, being a member of an ethnic minority, and a parental history of psychiatric disease or drug and alcohol abuse.57
There are many secondary causes of a ‘spasmus nutans-like syndrome’ which can be divided into neurological (vermian cerebellar hypoplasia, brain tumours, Pelizaeus Merzbacher disease, and Leigh's encephalomyelopathy) or ophthalmological origin (severe refractive errors, optic nerve and chiasmal gliomas, and retinal disorders).56 Importantly, the aforementioned conditions cannot always be excluded even in the absence of additional neurological, ophthalmological, or systemic findings or even with a full clinical resolution of the spasmus nutans-like symptoms.58 It is thus recommended that all patients with possible spasmus nutans have a brain MRI, visual-evoked potentials and electroretinography to exclude a potential underlying pathology.56
Shuddering attacks
Shuddering (or shivering) attacks are brief bursts of rapid, shivering-like movements of the head and both arms, occurring up to 100 episodes per day. They can begin in infancy or in older, otherwise healthy, children, and typically resolve over time. The attacks last several seconds, occasionally associated with stiffening of the upper extremities, without impairment of consciousness or loss of tone; features which aid in the differential diagnosis of seizures. However, shuddering attacks are usually misdiagnosed as tonic, myoclonic and infantile spasms. Additional conditions to be excluded are hyperekplexia and tics.59
The pathophysiology of shuddering attacks is unknown but a relationship with ET has been postulated. This was initially based on positive family history of ET in some of the affected children, the evolution to head tremor, the response to propranolol and due to a frequency similar to that of ET as seen on EMG.59 However, such a relationship has not been confirmed by other studies which did not find a history of shuddering attacks during infancy or among other family members in patients with ET as well as confirmed the long-term spontaneous remission of these attacks.59
Overall, shuddering attacks appear to be a benign movement disorder not requiring any treatment; however, propranolol may be considered in selected cases. Although these episodes are not associated with epileptiform activity on EEG, they were frequently misdiagnosed as seizures, thus resulting in unnecessary investigations or drug treatment.59
Conclusions
Tremor is a common and non-specific result of the malfunctioning of the nervous system and can also be seen in patients with a variety of acquired systemic conditions (eg, hypoxia) or structural lesions. Many other primary neurological diseases presenting with a more diffuse neurological impairment can also cause tremor (box 1), but in these instances, the occurrence of additional signs assist in the diagnostic process. As a general rule, most rare tremors present with an action tremor and a variable combination of postural and kinetic components. With few exceptions (eg, myorhythmia), resting tremors are more specifically related to the impairment of the central dopaminergic pathways and are less frequently seen.
Other medical conditions associated with tremor*
47,XYY (Klinefelter syndrome; Bardsley et al, 2013)
48, XXYY syndrome (Lote et al, 2013)
Alpers syndrome (Hunter et al, 2011)
Amyotrophic lateral sclerosis (Yavnai and Aizenbud, 2013)
Angelman syndrome (Thibert et al, 2013)
Antiphospholipid syndrome (Martino et al, 2006)
Bassen-Kornzweig syndrome (Soejima et al, 2006)
Behcet disease (Okuyucu et al, 2010)
Bing-Neel syndrome (Kolbaske et al, 2009)
Cabezas syndrome (Badura-Stronka et al, 2010)
Chronic subdural haematoma (Lin and Chang, 1997)
Crigler-Najjar syndrome (Perretti et al, 2007)
Cystathionine β-synthase deficiency (Wada et al, 2000)
Dentatorubral-pallidoluysian atrophy (Ohizumi et al, 2002)
Dravet syndrome (Genton et al, 2011)
Eosinophilia-myalgia syndrome (Kaufman, 1994) and hypereosinophilia (Yasui-Furukori et al, 2012)
Episodic ataxia type1 (Klein et al, 2004)
Fahr’s syndrome (El Hechmi et al, 2014)
Frontotemporal dementia (Curcio et al, 2002)
Galactosemia (Shah and Kuchhai, 2009)
Gaucher disease (Machaczka et al, 2012)
Gerstmann-Sträussler-Scheinker syndrome (Iwasaki et al, 2009)
Gillespie syndrome (Ticho et al, 2006)
Gliomatosis cerebri (Jayawant et al, 2001)
Hemiparkinsonism-hemiatrophy (Dziadkiewicz et al, 2013)
Hepatocerebral degeneration-like syndrome (Guler et al, 2013)
Hand-foot-mouth disease-associated brainstem encephalitis (enterovirus 71; Zeng et al, 2012)
High-pressure neurological syndrome (Jain, 1994)
Hirayama disease (Finsterer et al, 2013)
Homocystinuria (Varlibas et al, 2009)
Huntington's disease (especially the Westphal variant; Hefter et al, 1987)
Hypertrophic cranial pachymeningitis (Okimura et al, 1991)
HTLV-I-associated myelopathy (Puccioni-Sohler et al, 2005)
Intracranial hydatid disease (Onal et al, 2001)
Kabuki syndrome (Grunseich et al, 2010)
Kikuchi-Fujimoto disease (Moon et al, 2009)
L-mannosidosis (Szleper et al, 1991)
Langerhans cell histiocytosis (Nakamura et al, 2012)
Lissauer’s general paresis (Yamaguchi et al, 1995)
Lyme disease (Moser, 2011)
Maple syrup urine disease (Carecchio et al, 2011)
MELAS (Kovacs et al, 2006)
MERRF syndrome (Mancuso et al, 2013)
Metachromatic leucodystrophy (Duckett et al, 1975)
Methylmalonic acidemia (Ma et al, 2011)
Mohr-Tranebjaerg syndrome (Kim et al, 2007)
Nasu-Hakola disease (membranous lipodystrophy; Yokoi et al, 1988)
Nephronophthisis type 1 (Joubert syndrome-related disorders; Caridi et al, 2006)
Neuroferritinopathy (Kubota et al, 2009)
Paraneoplastic cerebellar syndrome (Mancuso et al, 2011)
Pearson syndrome (Lee et al, 2007)
Pelizaeus-Merzbacher disease (Kim et al, 2008)
Pontine and extrapontine myelinolysis (Aydin et al, 2003)
Primary central nervous system vasculitis (Araya et al, 2009)
Progressive myoclonic epilepsy type 1 (Kalviainen et al, 2008)
Reflex sympathetic dystrophy (Veldman et al, 1993)
Roussy-Lévy syndrome (Wake et al, 2000)
Sandifer’s syndrome (Demir et al, 2001)
Sneddon’s syndrome (Da Silva et al, 2005)
Spinal and bulbar muscular atrophy (eg, Kennedy syndrome; Finsterer, 2009)
Spontaneous intracranial hypotension (Turgut et al, 2010)
Stiff-man syndrome (Diniz et al, 1986)
Subacute spongiform encephalopathy (Kompoliti et al, 1999)
Superficial siderosis (Kresojevic et al, 2013)
Syringomyelia (Nogues et al, 1999)
Tick borne encephalitis (Duniewicz, 1976)
Tyrosine hydroxylase deficiency (Chi et al, 2012)
Vitamin B12 deficiency (Koussa et al, 2003)
Wernekink commissure syndrome (Liu et al, 2012)
X linked Kallmann’s syndrome (Mayston et al, 2001)
Xeroderma pigmentosum (Mimaki et al, 1986)
*First author and year of publication are listed in the table; see online supplementary material for a complete reference.
HTLV-I, human T cell lymphotropic virus type 1; MELAS: mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; MERRF, myoclonic epilepsy with ragged-red fibers.
The differential diagnosis of rare tremor syndromes should also include a few other conditions, especially epilepsy (epilepsia partialis continua) and myoclonus. Myoclonus is an involuntary movement due to a very fast and transient, usually shorter than 100 ms, muscular contraction (positive forms) or lapse in muscle tone (negative forms). Rhythmic myoclonus is rare, but in some cases, the regularity of jerks might confer a pseudorhythmic appearance.60 Therefore, some forms of pseudorhythmic myoclonus can be difficult to differentiate from tremor (table 4).
Pseudorhythmic myoclonus in the differential diagnosis of tremor
In conclusion, the aforementioned rare conditions should be considered in the differential diagnosis of various tremors, since the recognition of these entities will facilitate correct diagnoses and guide physicians to a prompt intervention in selected cases.
Acknowledgments
The authors would like to thank Dr Ali Rajput and Dr Eng-King Tan for providing us with the clinical data and the videos of their patients with myorhythmia.
References
Footnotes
Contributors RJU was involved in research project organization and execution, manuscript preparation and writing of the first draft. SD was involved in research project execution and manuscript preparation and review and critique. AEL was involved in research project conception and manuscript preparation and review and critique. AF was involved in research project conception and organization, and manuscript preparation and review and critique.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.