Article Text

Abstract
Objectives Neuromyelitis optica spectrum disorders (NMOSD) are inflammatory conditions of the central nervous system and an important differential diagnosis of multiple sclerosis (MS). Unlike MS, the course is usually relapsing, and it is unclear, if progressive neurodegeneration contributes to disability. Therefore, we aimed to investigate if progressive retinal neuroaxonal damage occurs in aquaporin4-antibody-seropositive NMOSD.
Methods Out of 157 patients with NMOSD screened, 94 eyes of 51 patients without optic neuritis (ON) during follow-up (F/U) and 56 eyes of 28 age-matched and sex-matched healthy controls (HC) were included (median F/U 2.3 years). The NMOSD cohort included 60 eyes without (EyeON −) and 34 eyes with a history of ON prior to enrolment (EyeON+). Peripapillary retinal nerve fibre layer thickness (pRNFL), fovea thickness (FT), volumes of the combined ganglion cell and inner plexiform layer (GCIP) and the inner nuclear layer (INL) and total macular volume (TMV) were acquired by optical coherence tomography (OCT).
Results At baseline, GCIP, FT and TMV were reduced in EyeON+ (GCIP p<2e−16; FT p=3.7e−4; TMV p=3.7e−12) and in EyeON − (GCIP p=0.002; FT p=0.040; TMV p=6.1e−6) compared with HC. Longitudinally, we observed GCIP thinning in EyeON− (p=0.044) but not in EyeON+. Seven patients had attacks during F/U; they presented pRNFL thickening compared with patients without attacks (p=0.003).
Conclusion This study clearly shows GCIP loss independent of ON attacks in aquaporin4-antibody-seropositive NMOSD. Potential explanations for progressive GCIP thinning include primary retinopathy, drug-induced neurodegeneration and retrograde neuroaxonal degeneration from lesions or optic neuropathy. pRNFL thickening in the patients presenting with attacks during F/U might be indicative of pRNFL susceptibility to inflammation.
Statistics from Altmetric.com
Introduction
Neuromyelitis optica spectrum disorders (NMOSD) are relapsing autoimmune inflammatory diseases of the central nervous system (CNS)1 and an important differential diagnosis of multiple sclerosis (MS), the most common autoimmune inflammatory disorder of the CNS.2 Unlike MS, which is thought to be a B-cell-mediated and T-cell-mediated demyelinating disease with early axonal pathology,3 NMOSD is considered a complement-dependent and antibody-mediated astrocytopathy, with 60%–80% of patients having detectable autoantibodies against the astrocytic water channel aquaporin-4 (AQP4-ab).1 4 5
Chronic neurodegeneration and disability progression during the progressive stage are observed in more than half of all patients with MS6 where there is no efficient treatment available.7 In contrast, patients with NMOSD virtually always present with monophasic or relapsing disease courses8 and it remains unclear, if progressive disease independent of clinical attacks also occurs in NMOSD.9 10
In 55% of patients with NMOSD, optic neuritis (ON) is the first clinical manifestation and frequently leads to severe structural damage of the afferent visual pathway with resulting functional impairment.11–13 The axon of the retinal ganglion cell and the ganglion cell itself are highly affected by ON and therefore are suitable targets to investigate neuroaxonal damage in NMOSD.14 Optical coherence tomography (OCT) is a non-invasive interferometric technique, which has been shown to provide high-resolution images of these retinal structures allowing an accurate quantification of their alterations.14
We and others recently found evidence for ON-independent retinal changes in NMOSD,15–17 which supports experimental data from animal models, suggesting an underlying astrocytopathy.10 18 Therefore, we aimed to investigate if progressive retinal neuroaxonal damage occurs in NMOSD. For this, we longitudinally followed AQP4-ab seropositive patients with NMOSD and healthy controls (HC) with OCT. We excluded all eyes with an acute ON during follow-up (F/U) and investigated eyes without previous ON and those with a history of ON separately to isolate ON independent effects.
Methods
Patients
One hundred and fifty-seven AQP4-ab seropositive patients with NMOSD from longitudinal observational cohort studies at the NeuroCure Clinical Research Center (NCRC) at Charité — Universitätsmedizin Berlin, Germany (EA1/131/09), the Ludwig Maximilians Universität Munich, Germany (Z427-14) and the Nuffield Center for Clinical Neurosciences at Oxford University, UK (REC 16/SC/0224) were screened for this study. Inclusion criteria were a definite diagnosis of AQP4-ab seropositive NMOSD according to 2015 international consensus criteria,19 complete longitudinal clinical and OCT imaging data with minimum F/U of 1 year, and age between 18 and 75 years at baseline. Eyes with ophthalmological comorbidities, insufficient OCT image quality, ON <5 months before F/U as well as patients with ON in both eyes during F/U were excluded. For comparison, we included longitudinal data of 28 matched HCs from the NCRC’s research database with equal median duration of F/U. High-contrast visual acuity (VA) was acquired under best correction in a subset of 22 patients with NMOSD from Berlin and Oxford (28 eyes without a history of ON, 11 eyes with a history of ON; F/U (median (min–max)) (1.1 (0.7–3.3)) with Early Treatment in Diabetes Retinopathy Study charts at 20 ft distance with retroilluminated charts. All participants gave written informed consent. The study was conducted in accordance with the current version of the Declaration of Helsinki and the applicable German and British laws.
Optical coherence tomography
All centres used Spectralis SD-OCT (Heidelberg Engineering, Heidelberg, Germany) with automatic real-time (ART) function for image averaging. The combined ganglion cell and inner plexiform layer volume (GCIP), inner nuclear layer volume (INL) and total macular volume (TMV) was calculated as a 3 mm diameter cylinder around the fovea from a macular volume scan (Berlin: 25°×30°, 61 vertical B-scans, 11≤ART≤18; Munich: 20°×20°, 25 vertical B-scans, 21≤ART≤49; Oxford: 69% 30°×25° 61 vertical B-scans, 27% 30°×15° 37 horizontal B-scans, 2% 30°×25° 61 horizontal B-scans, 2% 30°×15° 19 horizontal B-scans, 7≤ART≤22).20 The fovea thickness (FT) was defined as mean thickness of the 1 mm diameter cylinder around the fovea from the same scan.16 The peripapillary RNFL (pRNFL) was measured with activated eye tracker using 3.4 mm ring scans around the optic nerve (12°, 1536 A-scans, 1≤ART≤99) and the most inner 3.5 mm ring of a star-and-ring scan around the optic nerve (12°, 768 A-scans, 22≤ART≤32). Segmentation of all layers was performed semi-automatically using software provided by the OCT manufacturer (Eye Explorer 1.9.10.0 with viewing module 6.3.4.0, Heidelberg Engineering). One experienced rater (FCO) carefully checked all scans for sufficient quality and segmentation errors and corrected if necessary.21 OCT data in this study are reported according to the APOSTEL recommendations.22
Statistical methods
Group differences between NMOSD and HC were tested by Χ2 test for sex and Wilcoxon rank sum test for age. The primary outcome was the change of GCIP over F/U; secondary outcomes were changes in pRNFL, INL, FT and TMV. Cross-sectional differences of OCT values and VA between all groups were analysed pairwise by generalised estimating equation models to account for intereye within-patient correlations of monocular measurements. Longitudinal analysis of OCT and VA values was performed by the linear mixed-effects model: OCTvalue~time from baseline×group+(1+time from baseline|patient/eye)+(1|age)+(1|sex) and results are reported for (time from baseline×group). Marginal and conditional coefficients of determination for the models were estimated by pseudo-R2 for mixed-effect models. To exclude influences of contralateral ON, we included the contralateral ON status as random effect in a subanalysis. Annual loss was estimated for each individual as change to baseline at last visit divided by F/U time. For an exploratory subgroup analysis in patients with NMOSD with other, non-ON attacks during F/U, we defined the visit before first attack as new baseline. For VA analysis, we defined the first visit with best-corrected VA assessments as new baseline. All tests and graphical representations were performed with R V.3.3.1,23 with packages beeswarm, psych, geepack, ggplot, lme4, lmer, MuMIn, Rmisc and multcomp. Statistical significance was established at p<0.05.
Results
Cohort description and follow-up
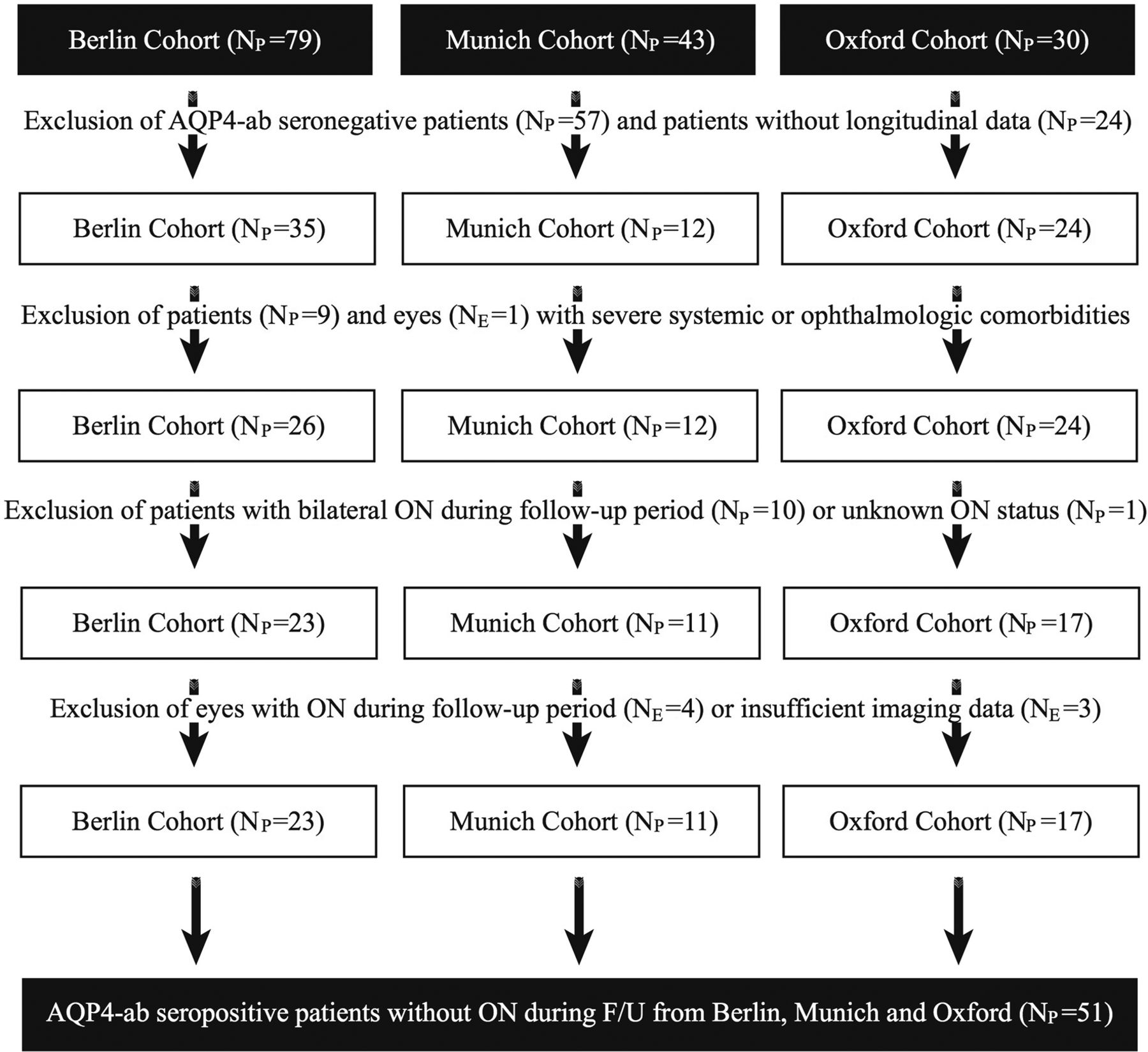
From the 157 patients with NMOSD screened, data from 51 patients with NMOSD (94 eyes) with a median F/U time of 2.3 years (range 1.0–3.5 years) from Berlin (n=23), Munich (n=11) and Oxford (n=17) fulfilled all inclusion criteria (figure 1 and table 1). The study cohort included 76% Caucasians, 10% African-Caribbeans, 8% Asians, 2% Middle Eastern, and 2% people of mixed origin; for 2% ethnicity was not available. Expanded Disability Status Scale assessment at baseline was available for 31 patients with NMOSD (table 1). Furthermore, we included OCT data from 28 age-matched and sex-matched HCs.
Demographic overview

NMOSD cohort selection.AQP4-ab, aquaporin-4 autoantibodies; F/U, follow-up; NMOSD, neuromyelitis optica spectrum disorders; NPnumber of patients; NE, number of eyes; ON, optic neuritis.
Group differences at baseline
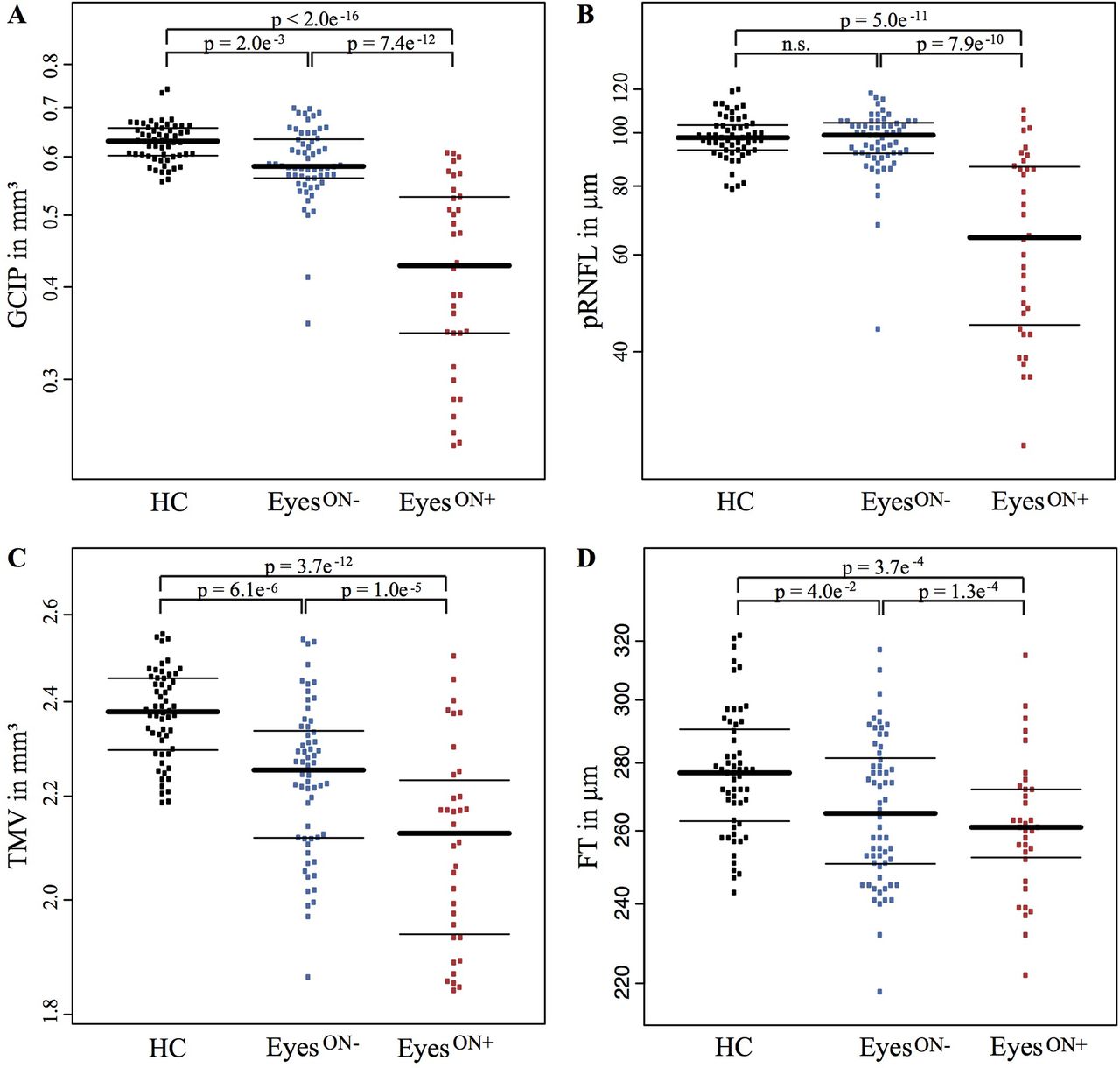
First, we analysed group differences at baseline between eyes from patients with NMOSD with history of ON (EyesON+), eyes from patients with NMOSD without previous ON (EyesON −) and eyes from HC. As expected, GCIP in EyesON+ was lower than in HC (p<2e−16), representing well-established ganglion cell damage after ON (table 2). GCIP in EyesON − was also lower than in HC eyes (p=0.002), but higher than in EyesON+ (p=7.4e−12) (figure 2A). Two eyes (3.3%) of EyeON − had GCIP values <2 SD of the mean GCIP of EyeON −. A subset of EyeON − having experienced ON neither in the ipsilateral nor in the contralateral eye also showed reduced GCIP (0.60±0.05 mm3) compared with HC (0.63±0.04 mm3, p=0.016). We found no pRNFL reduction in EyesON − in comparison with HC eyes, but as expected EyesON+ had reduced pRNFL as a marker of retinal axonal loss compared with EyesON − (p=7.9e−10) and HC (p=5.0e−11) (figure 2B). INL was comparable between the three groups. Also, TMV was reduced in EyesON+ (p=3.7e−12) and EyesON− (p=6.1e−6) compared with HC (figure 2C). EyesON − showed still higher TMV than EyesON+ (p=1.0e−5). FT was reduced in EyesON+ (p=3.7e−4) and again in EyesON − (p=0.040) in comparison with HC (figure 2D).

Beeswarm plots of cross-sectional OCT data for HC (black, left), NMOSD EyesON− (blue, middle) and NMOSD EyesON+ (red, right) (median±IQR, single eyes as dots) for (A) GCIP, (B) pRNFL, (C) TMV and (D) FT. EyesON −, patients with NMOSD without a history of ON; EyesON+, patients with NMOSD with a history of ON; FT, fovea thickness; GCIP, combined ganglion cell and inner plexiform layer; HC, healthy control; NMOSD, neuromyelitis optica spectrum disorders; n.s., not significant; OCT, optical coherence tomography; pRNFL, peripapillary retinal nerve fibre layer; TMV, total macular volume.
Baseline OCT results of patients with NMOSD and HCs.
OCT changes during follow-up
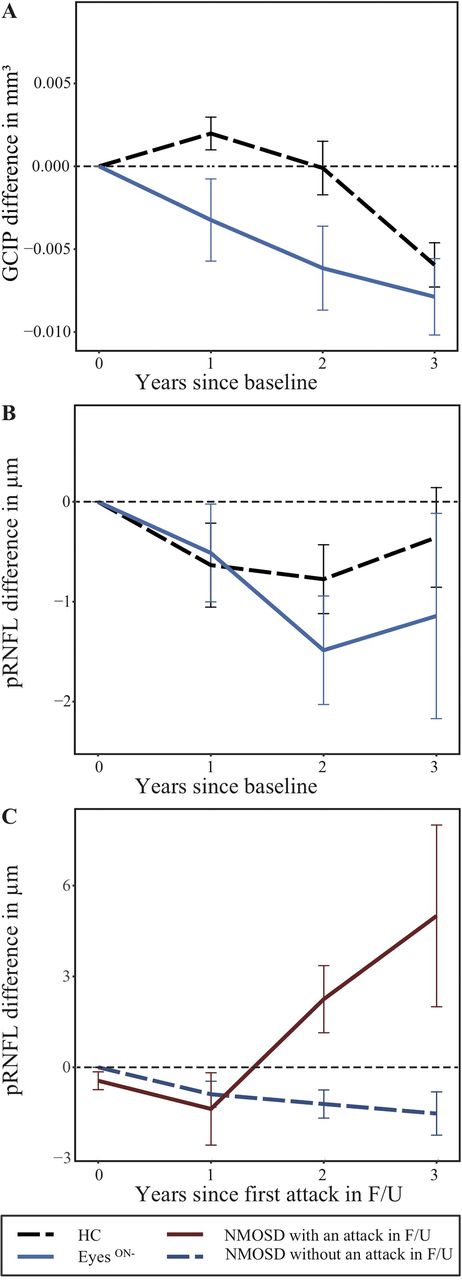
Longitudinally, we observed a thinning of GCIP in EyesON− (B=−0.004 SE=0.001 p=0.022; annual loss −0.00415±0.01200 mm3). This effect remained significant against HC (annual loss HC −0.00005±0.00466 mm3; p=0.044; figure 3). The longitudinal effect in EyeON− did not differ between eyes without a history of any ON (annual loss −0.00468±0.01310 mm3) compared with eyes with history of a contralateral ON before baseline (−0.00413±0.00779 mm3; p=0.805). EyesON+ revealed no longitudinal GCIP changes compared with HC (annual loss −0.00094±0.0119 mm3; p=0.960), which we interpret as a potential flooring effect resulting from previous ON episodes. pRNFL, TMV, FT and INL did not change in EyesON− or EyesON+ longitudinally compared with HCs (table 3). We excluded potential influences of ethnicity and treatment by incorporating respective factors in additional models (data not shown). In a subanalysis, GCIP change was not different between EyesON− treated with rituximab or azathioprine (p=0.710). Also, we excluded potential influences of contralateral ON in EyesON− (n=2) on longitudinal GCIP changes by including contralateral ON during F/U as additional factor, which led to the same p -value as without (p=0.044).
{kind=link}
{kind=link}
{kind=link}
Plots of longitudinal OCT data. Plotted change for rounded time since baseline for (A) GCIP and (B) pRNFL for EyesON− (blue, continuous) and HC (black, open-worked). EyesON+ are not shown due to high noise suggestive of a flooring effect. (C) pRNFL changes in patients with NMOSD with (dark red, continuous) and without (dark blue, open-worked) any attack during F/U independent from ON status. Line as connected means±SE. EyesON−, NMOSD eyes without a history of ON; F/U, follow-up; GCIP, combined ganglion cell and inner plexiform layer; HC, healthy control; NMOSD, neuromyelitis optica spectrum disorders; ON, optic neuritis; OCT, optical coherence tomography; pRNFL, peripapillary retinal nerve fibre layer.
Longitudinal OCT results of patients with NMOSD and HCs.
Seven patients with NMOSD (10 eyes) experienced attacks during F/U (two patients with one LETM, one patient with four LETMs, four patients (eyes) with contralateral ON). These patients showed pRNFL thickening (1.56±4.39 µm) compared with patients with NMOSD without any attacks during F/U (−1.04±3.21 µm; B=1.912 SE=0.597, p=0.003; figure 3), but no differences in GCIP (p=0.513), TMV (p=0.670), INL (p=0.970) or FT (p=0.330).
High-contrast visual acuity
At baseline, VA was not significantly lower in EyeON+ ((logMAR) 0.41±0.69) compared with EyeON− ((logMAR) 0.01±0.25, p=0.052). VA improved slightly in both groups longitudinally (EyeON− (logMAR) −0.02±0.07; EyeON+ (logMAR) −0.21±0.40) without discrepancy (p=0.054). VA of EyeON− was within normal range compared with published data.24
Discussion
We observed longitudinal GCIP loss in AQP4-ab seropositive patients with NMOSD without a history of ON. Using intraretinal layer segmentation and three-dimensional macular OCT, our results may be indicative of neuroaxonal damage both at baseline and progressively during F/U. In contrast, in eyes from patients with a new NMOSD-related attack, we did not observe pRNFL loss, but an increase in pRNFL thickness during F/U.
Cross-sectional retinal imaging studies in NMOSD have shown conflicting results as to whether inner retinal layer thinning can occur independently from ON in patients with NMOSD: While some studies reported reduced pRNFL or GCIP thickness in eyes without history of ON as potential evidence for ON-unrelated damage,15 25 26 others did not find differences compared with HC.11 27 28
We suggest that at least some of the reported neuroaxonal damage in NMOSD might be caused by a primary retinopathy. In the human retina, three types of astrocytic cells can be distinguished: (1) Müller cells, damage to which was previously suspected to cause foveal changes in NMOSD in cross-sectional and animal studies,15 16 29 30 (2) elongated retinal astrocytes are located in the RNFL, (3) whereas star-shaped astrocytes reside in the ganglion cell layer.31 Astrocytic dysfunction could lead to neuroaxonal damage in the retina and other affected brain regions, as has been reported in brainstem and chiasm, which was recently also shown in animal models of NMOSD.18 32 Microstructural changes have been reported in biopsies from spinal cord lesions,33 in spinal cord atrophy in AQP4-ab seropositive patients with NMOSD without previous myelitis34 and fovea thickness as well as optic radiation changes in AQP4-ab seropositive patients without previous ON16– all suggesting a primary astrocytopathy.
Besides a disease-related retinopathy, drug-induced neuroaxonal damage has to be considered. For example, it is well established that aggressive immunosuppressive treatment by cytostatic or cytotoxic drugs as well as methylprednisolone might induce neuronal damage.35 Patients with NMOSD are regularly on these treatments, and methylprednisolone still presents the primary option for treating acute attacks. In an exploratory analysis comparing patients on rituximab treatment versus those on azathioprine, we did not detect any differences. However, our study was most likely underpowered to investigate drug-related changes.
Alternatively, retrograde neuroaxonal degeneration has to be considered as an explanation for GCIP thinning.36 In NMOSD, ON often spans several segments of the optic nerve, and chiasmal crossover is reported in some cases.37 In our study, 3.3% of eyes without history of ON showed severely reduced GCIP at baseline, indicative of chiasmal affection and crossing-over during acute ON, which was clinically apparent only unilaterally. On the other hand, also patients who never experienced ON showed decreased GCIP compared with HC at baseline pointing towards a pathology independent from bygone optic nerve damage. Although, subclinical ONs, which might have occurred before the study, could be considered. Both are, however, unable to explain the observed GCIP loss during F/U: first, we accounted for contralateral ONs in F/U. And second, we did not observe pRNFL or VA loss that go along with GCIP thinning. It should be kept in mind that this study did not account for potential retrograde degeneration originating from posterior visual pathway damage, which is mandatory for future confirmatory studies.
Interesting is our exploratory finding of a mild pRNFL thickening in the few patients presenting with a NMOSD-related attack during F/U. Although this should not be overstated in light of the small sample size, it could indicate that astrocytes in regions not directly affected by an acute lesion or attack could be affected during an attack elsewhere in the CNS. But a mildly swollen pRNFL might also be an effect of prednisolone treatment in acute state or covert subclinical axonal damage in the pRNFL in line with an underlying optic neuropathy, which limits certain conclusions in this context. This finding might also explain conflicting results of two previous longitudinal studies: Manogaran et al did not find significant pRNFL or macular thinning in a case series of nine patients with NMOSD from Canada and two, 4 years apart visits.38 Bouyon et al reported pRNFL thinning over 18 months in 30 patients with NMOSD from France.39
Given the rarity of NMOSD in Europe, an important strength of our study is the large sample size.40 Furthermore, we were able to distinguish disease-related changes from physiological changes by including a matched HC cohort. We also thoroughly excluded potential confounders like AQP4-ab seronegative patients, or patients within 5 months after an acute ON, in which ongoing neuroaxonal degeneration from ON might still be present. Important limitations of our study include the lack of consistent longitudinal functional measurements in HCs and sensitive functional tests, which prohibit investigating the functional relevance of these changes, and the lack of HCs from all study-sites. Furthermore, our study cannot conclusively investigate the influence of disease-modifying therapy, ethnicity and contralateral ON because of limited sample size for these subgroup analyses.
Our findings, if confirmed, challenge the notion that disease-related damage occurs only attack dependent in NMOSD, which is the hallmark of today’s immunosuppressive therapy of NMOSD. If confirmed, our data suggest that targeting a progressive retinopathy might be important for monitoring and treatment of the disease. The clinical relevance of these changes still needs to be investigated. It also remains to be shown, if similar observations can be made also in other areas of the CNS.
Acknowledgments
The authors would like to thank Dr Janine Mikolajczak, Charlotte Bereuter, Jan Lödige and Ivonne Hinz for their excellent technical support. SJ would like to thank Mrs Anna Eschlbeck and the Nikon Imaging Center at the University of Heidelberg for excellent technical assistance.
References
Footnotes
FP and AUB are joint senior authors.
FCO and JH are joint first authors.
Contributors FCO: data collection, OCT data processing, data analysis, writing of the manuscript. JH: data acquisition and collection, study coordination. ARF: data collection. NL: data collection, data analysis. HZ: data acquisition, study coordination, supervision of OCT data processing. SM: data analysis. NB/JBS/JP/MIL/TK: data acquisition, study coordination. OBW/PA/KR: study coordination. SJ: data acquisition. FP: data acquisition, study coordination, study concept. AUB: study concept, design, study coordination, data analysis, writing of the manuscript. All authors revised the manuscript for intellectual content and read and approved the final manuscript.
Funding The project was supported with grants from the German Ministry for Education and Research (BMBF/KKNMS; Competence Network Multiple Sclerosis (to FP, KR), from the Deutsche Forschungsgemeinschaft (DFG, grant Exc. 257 to FP, AUB), from the German Federal Ministry of Economic Affairs and Energy (EXIST 03EFEBE079 to AUB), from the German Ministry of Education and Research (N2-ADVISIMS 16GW0079 to FP and AUB), from the National Multiple Sclerosis Society (to FP), from the Guthy-Jackson Charitable Foundation (to FP, AUB) and from Novartis (to HZ). MIL and JP were funded for highly specialised service for neuromyelitis optica—NHS, UK, grants. JP also received research support from the MS society and Guthy-Jackson Charitable Foundation. FCO received financial support by the MD student research stipend from the Berlin Institute of Health (BIH).
Competing interests FCO has nothing to disclose. JH reports personal fees and non-financial support from Merck, grants, personal fees and non-financial support from Novartis, personal fees from Roche, non-financial support from Bayer Healthcare, personal fees from Santhera, personal fees and non-financial support from Biogen, personal fees and non-financial support from Sanofi Genzyme, all outside the submitted work. ARF is sponsored by Abide Therapeutic outside of the submitted work and reports no potential conflicts of interest. NL has nothing to disclose. HZ reports grants from Novartis, during the conduct of the study. SM has nothing to disclose; he is named as inventor on a patent application describing OCT image analysis. NB has nothing to disclose. JBS received speaking fees and travel grants from Bayer Healthcare, Sanofi-Aventis/Genzyme, Biogen and Teva Pharmaceuticals, unrelated to the present scientific work. PA reports grants, personal fees and non-financial support from Allergan, personal fees and non-financial support from Bayer Healthcare, grants, personal fees and non-financial support from Biogen, grants, personal fees and non-financial support from Ipsen, grants, personal fees and non-financial support from Merz Pharmaceuticals, personal fees and non-financial support from Merck, grants, personal fees and non-financial support from Novartis, non-financial support from Sanofi-Aventis/Genzyme, personal fees and non-financial support from Teva Pharmaceuticals, grants, personal fees and non-financial support from Roche, outside the submitted work. KR reports research support from Novartis and Merck Serono as well as speaking fees or travel grants from Bayer Healthcare, Biogen Idec, Merck Serono, Sanofi-Aventis/Genzyme, Teva Pharmaceuticals, Roche, Novartis and the Guthy-Jackson Charitable Foundation, all unrelated to this work. TK received travel expenses and personal compensations from Bayer Healthcare, Teva Pharmaceuticals, Merck, Novartis Pharma, Sanofi-Aventis/Genzyme, Roche and Biogen as well as grant support from Bayer-Schering AG, Novartis and Chugai Pharma, unrelated to this work. OW has nothing to disclose. SJ has nothing to disclose. MIL reports personal fees from Biogen Idec and grants from Novartis, outside the submitted work. JP reports grants Journal of Neurology, Neurosurgery and Psychiatry and personal fees from Merck Serono, grants and personal fees from Biogen Idec, personal fees from Teva Pharmaceuticals, grants from Chugai, grants and personal fees from MedImmun, grants and personal fees from Alexion, grants and personal fees from Novartis, personal fees from Roche, grants from Genzyme, grants and personal fees from ABIDE, personal fees from TG Therapeutics, outside the submitted work. In addition, JP has a patent Isis: Diagnosing Multiple Sclerosis. FP reports research grants and speaker honoraria from Bayer, Teva, Genzyme, Merck, Novartis, MedImmune and is member of the steering committee of the OCTIMS study (Novartis), all unrelated to this work. AUB is cofounder and holds shares of commercial entities Motognosis and Nocturne. He is named as inventor on several patent applications describing serum biomarkers for MS, perceptive visual computing for tracking of motor dysfunction and OCT image analysis.
Patient consent Not required.
Ethics approval NeuroCure Clinical Research Center (NCRC) at Charité — Universitätsmedizin Berlin, Germany (EA1/131/09), the Ludwig Maximilians Universität Munich, Germany (Z427-
14) and the Nuffield Center for Clinical Neurosciences at Oxford University, UK (REC 16/SC/0224).
Provenance and peer review Not commissioned; externally peer reviewed.