

Abstract
One important therapeutic goal during CNS injury from trauma or demyelinating diseases such as multiple sclerosis is to develop methods to promote remyelination. We tested the hypothesis that spontaneous remyelination in the toxic nonimmune model of lysolecithin-induced demyelination can be enhanced by manipulating the inflammatory response. In PBS-treated SJL/J mice, the number of remyelinating axons per square millimeter of lesion area increased significantly 3 and 5 weeks after lysolecithin injection in the spinal cord. However, methylprednisolone or a monoclonal antibody (mAb), SCH94.03, developed for its ability to promote remyelination in the Theiler’s virus murine model of demyelination, further increased the number of remyelinating axons per lesion area at 3 weeks by a factor of 2.6 and 1.9, respectively, but did not increase the ratio of myelin sheath thickness to axon diameter or the number of cells incorporating tritiated thymidine in the lesion. After 3 weeks, the number of remyelinating axons in the methylprednisolone or mAb SCH94.03 treatment groups was similar to the spontaneous remyelination in the 5 week PBS control-treated group, indicating that these treatments promoted remyelination by increasing its rate rather than its extent. To address a mechanism for promoting remyelination, through an effect on scavenger function, we assessed morphometrically the number of macrophages in lesions after methylprednisolone and mAb SCH94.03 treatment. Methylprednisolone reduced the number of macrophages, but SCH94.03 did not, although both enhanced remyelination. This study supports the hypothesis that even in toxic nonprimary immune demyelination, manipulating the inflammatory response is a benefit in myelin repair.
Three general categories of experimental CNS demyelination can be distinguished: (1) toxin-induced [cuprizone (Blakemore, 1973), ethidium bromide (Yajima and Suzuki, 1979), and lysolecithin (Hall, 1972)]; (2) autoimmune-induced [experimental autoimmune encephalomyelitis (EAE) (Raine and Traugott, 1985)]; and (3) virus-induced [corona virus (Herndon, 1977) and Theiler’s murine encephalomyelitis virus (TMEV) (Rodriguez and Lennon, 1990)]. Spontaneous CNS remyelination has been observed in each of these models of demyelination in rodents. Some CNS lesions of multiple sclerosis (MS) in humans are remyelinated by oligodendrocytes (Prineas and Connell, 1979; Rodriguez and Sheithauer, 1994) or Schwann cells (Ghatak et al., 1973), although most lesions examined at autopsy show extensive demyelination and gliosis without myelin repair. In addition, demyelination and remyelination have been observed after acute spinal cord compression (Gledhill et al., 1973). These observations suggest that there is the potential for complete remyelination after demyelination in spinal cord injury or multiple sclerosis, but there are factors that prevent the full expression.
Two hypotheses have been proposed to explain the absence of full remyelination in the human CNS (Rodriguez and Lindsley, 1992). The absence of significant spontaneous remyelination may be attributable to either the presence of inhibitory elements preventing myelin repair or the absence of cells or factors necessary for new myelin synthesis. In the experimental model of demyelination induced by TMEV, a natural enteric picornavirus in mice, CNS remyelination can be promoted either by immunosuppression (Rodriguez and Lindsley, 1992) or by immunostimulation by passive transfer of Igs such as CNS-specific antiserum (Rodriguez et al., 1987), purified Ig (Rodriguez and Lennon, 1990), polyclonal mouse IgG (van Engelen et al., 1995), or a monoclonal autoantibody (mAb) designated SCH94.03 (Miller et al., 1994). The purpose of these experiments was to determine in a toxic nonimmune model of demyelination whether corticosteroids suppress inhibitory elements for myelin repair, which would allow for increasing the rate of spontaneous remyelination, and whether stimulation of the humoral immune response by administration of polyclonal or monoclonal antibodies provides factors for new myelin synthesis.
MATERIALS AND METHODS
Mice. Forty-six 12-week-old SJL/J (H-2s) mice weighing 20–25 gm were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were housed in plastic cages, and food and water were provided ad libitum. Handling of all animals conformed to the National Institute of Health and Mayo Clinic institutional guidelines.
Surgery. Mice were anesthetized by intraperitoneal injection of sodium pentabarbitol (0.08 mg/gm). Dorsal laminectomies were performed on the upper thoracic region of the spinal cord. A 34 gauge needle attached to a Hamilton syringe mounted on a stereotactic micromanipulator was used to inject 1 μl of a 1% solution of lysolecithin (l-α-lysophosphatidylcholine) (Sigma, St. Louis, MO) in sterile PBS, pH 7.4, with Evan’s blue added as a marker. The needle was inserted into the anterior or lateral part of the spinal cord, lysolecithin solution was injected, and then the needle was slowly withdrawn. The wound was sutured in two layers, and mice were allowed to recover. The day of lysolecithin injection was designated day 0.
Treatments. Mice were assigned randomly to groups (four to nine animals per group) to receive the following treatments and were killed on days 14 (n = 6), 21 (n = 34), and 35 (n = 6) after lysolecithin injection. All mice were 12 weeks of age to exclude the potential bias of age on remyelination after demyelination (Gilson and Blakemore, 1993).
Methylprednisolone. Mice were treated with pulse doses of methylprednisolone (Depo-Medrol, 80 mg/ml; Upjohn, Kalamazoo, MI) given by intraperitoneal injections of 1 mg (∼45 mg/kg) on days 0, 3, 7, 10, 14, and 17 to determine whether steroids would enhance remyelination. This approach was used to test whether inhibition of the inflammatory response would enhance myelin repair. This approach also simulated treatments used in spinal cord injury (Bracken et al., 1990).
Polyclonal IgG. On days 7, 10, 14, and 17, mice were injected intraperitoneally twice daily with 0.5 mg of polyclonal IgG obtained from multiple mouse donors (1 mg/ml in PBS from Sigma lot 033H8860). This approach was identical to treatments used in other murine models in which IgG had been shown to promote remyelination (van Engelen et al., 1995). This approach also simulated the use of intravenous Ig (IvIg), which has been shown to be beneficial in a subset of patients with multiple sclerosis (Fazekas et al., 1997).
Anti-SCH Ig.Polyclonal Ig directed against spinal cord homogenate (anti-SCH Ig) was shown to enhance remyelination in the TMEV model of demyelination (Rodriguez et al. 1987, 1990). This was injected intraperitoneally (0.5 ml of 1.1 mg/ml solution) on days 7, 10, 14, and 17.
mAb SCH94.03 IgM-κ. A monoclonal antibody developed in our laboratory for its ability to promote remyelination in the TMEV model (Miller and Rodriguez, 1995) was injected intraperitoneally (0.1 mg) on days 7, 10, 14, and 17.
PBS. Control mice were given intraperitoneal injections of 0.5 ml of PBS on days 7, 10, 14, and 17. Three groups of mice were killed on days 14, 21, and 35 after lysolecithin injection to address the normal temporal profile of spontaneous remyelination in the lysolecithin model.
Histopathology. On days 14, 21, and 35, mice were killed for pathological analysis. After anesthesia with sodium pentobarbital, mice were perfused with Trump’s fixative (phosphate-buffered 4% formaldehyde containing 1% glutaraldehyde, pH 7.4). Spinal columns were removed and allowed to post-fix for 1–3 d until spinal cords were removed. Six to eight 1 mm coronal blocks were cut from the site marked by the Evan’s blue marker. This assured that the entire lesion area was examined. Serial blocks were kept in 24-well plates and washed with 0.1 m phosphate buffer. Blocks were secondarily fixed with osmium tetroxide and dehydrated through a graded alcohol series, washed in propylene oxide and embedded in Araldite (Polysciences, Warrington, PA). One-micrometer sections were cut and stained with 4% paraphenylenediamine. Selected areas were trimmed and prepared for electron microscopy. Ultrathin sections were placed on celloidin-coated 200-mesh grids, stained with 4% uranyl acetate in 50% methanol and counterstained with lead citrate. The grids were examined with a JEOL 1200 electron microscope.
In vivo analysis of [3H]thymidine incorporation. To detect proliferating cells, 40 animals were injected intraperitoneally with 100 μCi of [3H]thymidine (Amersham, Arlington Heights, IL) 48, 36, 24, and 12 hr before killing on days 14 (n = 5), 21 (n = 29), and 35 (n = 6). One-micrometer sections of spinal cord from mice previously injected intraperitoneally with [3H]thymidine were dipped in autoradiography emulsion (NTB2; Eastman Kodak, Rochester, NY), sealed in black boxes, and exposed for 3 weeks at 4°C. Slides were developed in Kodak D19 developer, rinsed in a distilled water stop bath, and fixed with Kodak fixer. Only lesion areas that were at least 0.02 mm2 in total area were studied morphometrically to exclude the possibility that very small lesions would undergo more rapid and extensive repair. Based on this assessment only five lesion areas from 40 mice with lysolecithin-induced lesions were excluded from the analysis.
Analysis of light and electron microscopic spinal cord section images. A Zeiss (Oberkochen, Germany) interactive digital analysis system attached to a Zeiss photomicroscope was used to measure the area (square millimeters) of each lysolecithin lesion from every block. The lesion was defined as the area that included axons that were completely and partially demyelinated as well as completely and partially remyelinated (Fig. 1). The lesion areas for each of the 35 mice killed at days 14, 21, and 35 were summed to provide the total lesion area per mouse. This system was also used to count the number of remyelinated axons in each lesion. Abnormally thin myelin sheaths, relative to axonal diameter, were the criterion for oligodendrocyte remyelination. Schwann cell remyelination was characterized by thicker myelin sheaths, a one cell per axon relationship, and a surrounding basement membrane. A cursor was used to mark each remyelinated axon (oligodendrocyte or Schwann cell) with a cross, and these were summed to determine the total number of remyelinated axons per total lesion area.

Demonstration of the methodology for determining lesion area measurements. A, Lysolecithin lesion from a PBS-treated control mouse 3 weeks after lysolecithin injection.B, Area in blue shows the normal white matter surrounding lesion that was used to discriminate lesion.C, Area in red demonstrates the area used in calculating the total lesion area.
Lysolecithin lesion areas were also viewed on a JEOL 1200 electron microscope. Areas of demyelination and oligodendrocyte-type remyelination were photographed. Photo negatives were incorporated into digital image files. These images were analyzed by the Zeiss digital analysis system to determine the ratio of myelin thickness to axon diameter.
In vivo analysis of macrophage infiltration. A Zeiss interactive image analysis system attached to a Zeiss photomicroscope was used to measure areas of phagocytic macrophage infiltrates in 41 lysolecithin lesion areas from mice killed at days 14, 21, and 35 (Fig.2). Macrophages were readily identified in the lesions as cells with extensive myelin debris-containing vacuoles. Because in certain areas macrophage infiltrates were very dense and individual macrophages could not be distinguished, cumulative areas of macrophage infiltrate were used to calculate the number of macrophages per area of lesion. The total area of infiltration was divided by the mean area of one macrophage (0.00127 mm2) to yield total macrophages. The data were expressed as number of macrophages per square millimeter of lesion area.

Demonstration of the methodology for macrophage enumeration. A, Region of macrophage infiltration that does not allow for distinction of individual cells. B, Region of macrophages from A discriminated to calculate total macrophage area. C, Region where individual macrophages can be distinguished. D, Region discriminated from C used to calculate individual size of macrophages. Note examples of axons remyelinated by Schwann cells (arrows).
Statistical analysis. Analyses were performed in a blinded manner on coded slides. Pathological abnormalities were scored without knowledge of treatment groups. Differences between treatment groups were analyzed using Kruskal–Wallis one-way ANOVA, and pairwise multiple comparisons were done using Dunn’s method (p < 0.05). Data that were normally distributed were analyzed by one-way ANOVA, and pairwise multiple comparisons were done by Dunnett’s method (p < 0.05).
RESULTS
Demyelination and remyelination in PBS control-treated SJL/J mice
Light and electron microscopy in the PBS-treated animals revealed focal areas of demyelination, invading macrophages laden with myelin debris, and thinly myelinated axons at 14, 21, and 35 days after lysolecithin injection (Fig.3A). No differences in lesion areas were found between the PBS-treated groups; the mean area ± SEM of lesion for these groups was 0.0709 ± 0.015 mm2(n = 17). Few if any lymphocytes were identified in lesions, as would be expected from toxin-induced demyelination. In previous pilot experiments we found that spontaneous oligodendrocyte remyelination, as detected by abnormally thin myelin sheaths in relation to axon diameter, started ∼1 week after lysolecithin injection, as has been reported in the mouse (Jeffery and Blakemore, 1995) and rat (Blakemore, 1976) previously. Most animals also had some evidence of PNS-type remyelination, characterized by the one-to-one relationship between CNS axons and Schwann cells, and a basement membrane surrounding newly myelinated axons. Five weeks after lysolecithin injection the total number of remyelinated axons per square millimeter of lesion area increased significantly compared with the 2 and 3 week groups (Table 1). The number of remyelinated axons per square millimeter increased 2.6-fold from 2 to 3 weeks and 6.2-fold from 2 to 5 weeks (Table 1). Myelin sheath thickness in axons remyelinated by oligodendrocytes also increased spontaneously from 2 to 5 weeks (Table 1, Fig.4) but never reached the thickness observed in normal axons.

Spinal cord sections from SJL/J mice injected with lysolecithin and treated with A, PBS control;B, methylprednisolone; C, mAb 94.03; orD, polyclonal Ig directed against spinal cord homogenate (SCH/Ig). Treatment was begun on day 0 (methylprednisolone) or day 7 (PBS control, mAb 94.03 or SCH/Ig) and continued until day 21 at time of kill. Note focal area of demyelination with dense macrophage infiltration with minimal remyelination in the mouse treated with PBS control (A). In contrast, there is oligodendrocyte remyelination, characterized by abnormally thin myelin sheath relative to axon diameter, and Schwann cell remyelination in mice treated with methylprednisolone (B), mAb SCH94.03 (C) or SCH/Ig (D). The number of remyelinated axons from sections such as these are shown in Tables 1 and 2 (× 750).
Spontaneous remyelination in PBS-treated SJL/J mice after lysolecithin injection in the spinal cord

Relative myelin sheath thickness as a function of axon diameter in axons remyelinated by oligodendrocytes at 2, 3 or 5 weeks in PBS-treated SJL/J mice injected with lysolecithin.
Demyelination and remyelination in Ig- or methylprednisolone-treated animals
At 3 weeks after lysolecithin injection, the total lysolecithin lesion area was 0.098 ± 0.01 mm2 (mean ± SEM), based on the analysis of the 23 mice shown in Table2. There was no statistical difference in the lesion areas when comparing different treatment groups. In contrast, there was a continuous increase in the number of remyelinating axons per square millimeter of lesion area from the lowest in the PBS control group, increasing in the polyclonal IgG, anti-SCH Ig, and mAb SCH94.03 to the highest in the methylprednisone group (Table 2).
Effects of corticosteroids and Ig treatment on remyelination 3 weeks after lysolecithin-induced demyelination in the spinal cord
Polyclonal IgG and anti-SCH Ig did not statistically improve remyelination, whereas mAb SCH94.03 and methylprednisolone treatment statistically increased the number of remyelinated axons per square millimeter of lesion area at the 3 week time point by a factor of 1.9 and 2.6, respectively (Table 2, Fig. 3). After 3 weeks the total number of remyelinated axons per square millimeter in the mAb SCH94.03 or methylprednisolone treatment groups (Table 2) was comparable with the spontaneous remyelination observed in the 5 week PBS control group (Table 1). The various treatments did not increase the ratio of myelin sheath thickness to axon diameter in fibers undergoing oligodendrocyte-type remyelination (Table 2).
In vivo analysis of [3H]thymidine incorporation
Although the number of cells incorporating [3H]thymidine decreased from 2 to 5 weeks after lysolecithin injection, no statistical differences were observed in the PBS control groups (Fig. 5). Furthermore, the number of cells incorporating [3H]thymidine in the various treatments did not differ statistically from the 3 week PBS control group (Fig. 5). The greatest number of proliferating cells was observed in mice receiving poly-IgG, but this was not statistically significant (Fig. 5). Although corticosteroids are potent antiproliferative agents for macrophages and other inflammatory cells, cells incorporating [3H]thymidine (presumed glial cells) were observed in mice receiving this treatment. Some [3H]thymidine-positive cells were identified as inflammatory (i.e., surrounding blood vessels or infiltrating the spinal cord parenchyma from the meninges). A few proliferating Schwann cells were observed near the root entry zone associated with PNS-type remyelination. Many of the [3H]thymidine-positive noninflammatory cells were presumptive glia near areas of oligodendrocyte remyelination. As described previously (Rodriguez and Lindsley, 1992), these cells had large nuclei without a definite nucleolus, a non-electron-dense cytoplasm, and no distinct glial fibrils and were usually at a distance from dense inflammatory infiltrates.

Number of proliferating cells per mm2 of lesion in PBS-treated groups and treatment groups at 3 weeks after lysolecithin injection (data expressed as proliferating cells per square millimeter of lesion + SEM).n, Number of mice studied.
In vivo analysis of macrophage infiltration
One potential mechanism for enhancing remyelination is by effecting the recruitment or function of scavenger macrophages in the lesion. Removal of myelin debris from the lesion could then allow progenitor glia to proliferate and differentiate to initiate myelination. We first assessed the number of macrophages in lysolecithin lesions induced at 2, 3, and 5 weeks before killing. The number of macrophages/mm2 of lesion for PBS-treated mice injected with lysolecithin was 731 ± 79 (n = 6) at 2 weeks (mean ± SEM), 1044 ± 185 at 3 weeks, and 643 ± 69 (n = 6) at 5 weeks. Although the number of macrophages appeared to peak at 3 weeks, there was no statistical difference in the data. To test the hypothesis that a change in the macrophage infiltrate would correlate with enhanced remyelination, we analyzed the number of macrophages per square millimeter in treatment groups at the 3 week time point. There was a statistically significant difference between the PBS-treated group and the methylprednisolone group by Dunnett’s multiple comparison procedure (Fig.6). Of interest, however, no decrease in the number of macrophages per lesion area was observed in the 94.03-treated group, which statistically enhanced remyelination, or in the anti-SCH Ig and poly-IgG groups, which did not significantly enhance remyelination. This result indicates that the mechanism of enhanced remyelination between methylprednisolone and 94.03 may be fundamentally different.
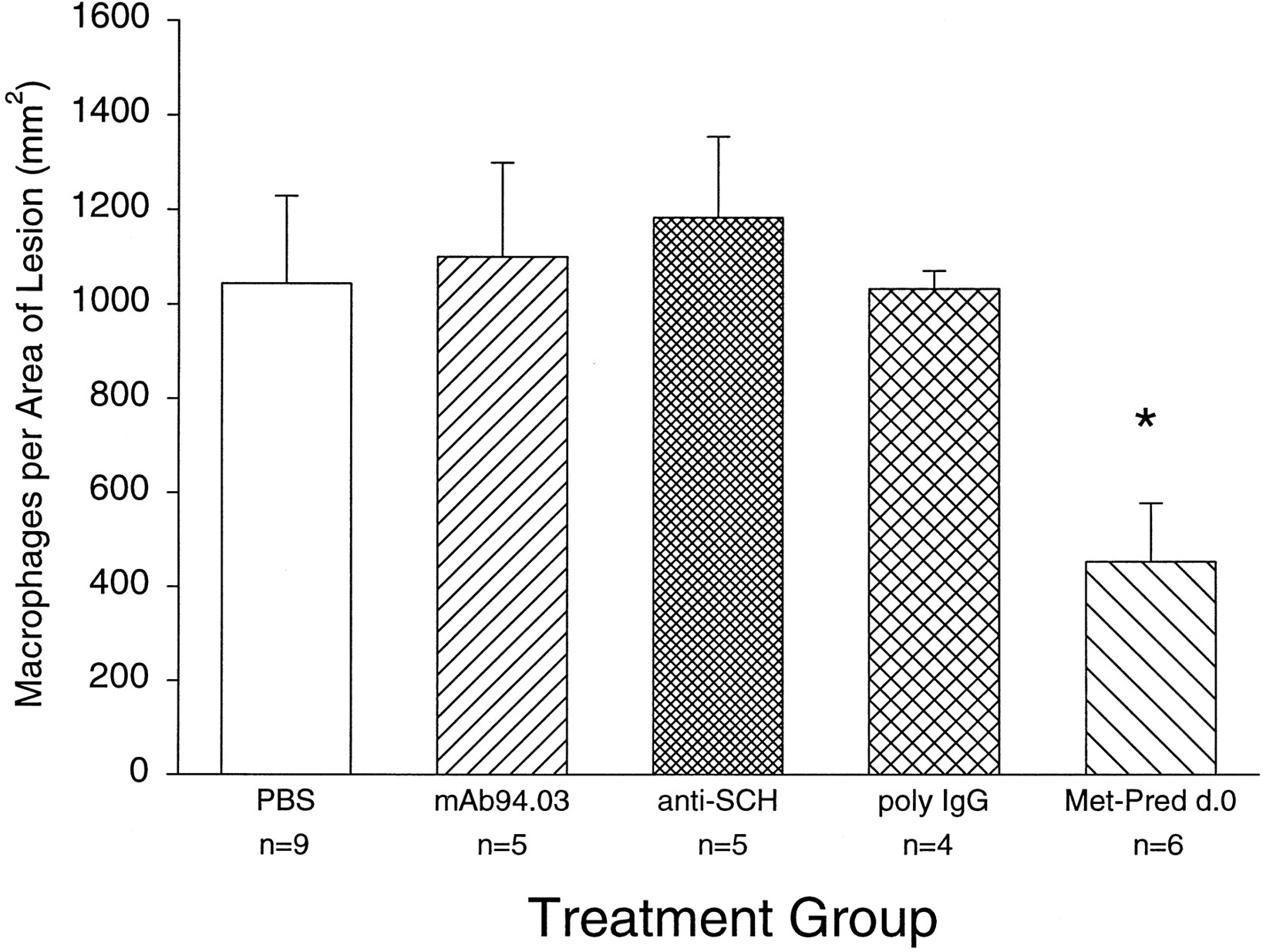
Number of macrophages per lesion area in treatment groups (data expressed as macrophages per square millimeter of lesion + SEM). n, Number of mice studied. Data marked by * denote statistical significance by Dunnett’s multiple comparison procedure with the PBS-treated group serving as the control.
DISCUSSION
This study demonstrated that spontaneous remyelination in a toxic–traumatic model of demyelination is a normal physiological response that can be promoted either by treatment with corticosteroids or by immunostimulation using mAb SCH94.03. These findings are comparable with experiments published previously in an immune-mediated virus model of demyelination, in which immunosuppression (Rodriguez and Lindsley, 1992) or mAb SCH94.03 (Miller et al., 1994) was able to promote CNS-type remyelination. Although there may be differences in the role of the inflammatory response in the genesis of toxic–traumatic versus virus-induced demyelination, the present study showed that a beneficial therapeutic effect by agents that potentially influence inflammation is not restricted to immune-mediated demyelination.
Theoretically a beneficial therapeutic effect could be caused either by decreasing demyelination or by increasing remyelination. Corticosteroid treatment could have been beneficial by inhibiting directly lysolecithin-induced demyelination, as reported by Triarhou and Herndon (1986). Lysolecithin injection causes primary myelin breakdown by increasing phospholipase A2 activity contained in activated macrophages (Trotter and Smith, 1986), which degrades membrane phosphatidylcholine into lysolecithin (Weltzien, 1979). Because corticosteroids inhibit phospholipase A2 (Crompton et al., 1988), they could counteract directly the demyelinating effect of lysolecithin on phospholipase A2. However, we found that corticosteroid administered within 0.5–1 hr after lysolecithin injection did not significantly decrease the lesion area. This is in contrast with the report of Triarhou and Herndon (1986) on dexamethasone acetate administration in rats, but the latter study did not specify the interval between lysolecithin injection and corticosteroid administration. In patients with acute spinal cord injury, treatment with methylprednisolone (as a bolus of 30 mg/kg followed by 5.4 mg · kg−1 · hr−1 for 23 hr) improves neurological recovery when the medication is given within the first 8 hr (Bracken et al., 1990), possibly by preventing the breakdown of membranes (Braughler and Hall, 1983). In our lysolecithin experiments, methylprednisolone was administered for 17 d instead of 23 hr as in the human trial (Bracken et al., 1990). Our results raise the possibility that the therapeutic effect of methylprednisolone in toxic or traumatic spinal cord injury may in part be the result of enhanced remyelination.
Two potential mechanisms exist by which treatment may promote spontaneous remyelination (Miller et al., 1994). First, some pathogenic component preventing full remyelination may be inhibited. If the outcome of the demyelinating process is based on a balance between tissue destruction and repair, inhibiting those components that prevent healing would allow a physiological repair response to predominate. Thus, corticosteroids may have promoted spontaneous CNS remyelination by their anti-inflammatory effects; i.e., perturbation of leukocyte trafficking, lytic action on lymphocytes, and inhibition of T-lymphocyte activation (Cupps and Fauci, 1982), or by interfering with locally secreted cytokines and lymphokines of interest. In these experiments the number of macrophages infiltrating the lesion at 3 weeks was significantly reduced in the methylprednisolone-treated group, which is consistent with the first hypothesis. Of interest, methylprednisolone has also been shown to inhibit inflammatory processes in experimental spinal cord lesions in the rat (Bartholdi and Schwab, 1995). Corticosteroids also directly effect lymphokine production, especially interleukin 1 (Lew et al., 1988), and interleukin-2 (IL-2) (Arya et al., 1984; Boumpas et al., 1991). IL-2 has been shown to stimulate (Benveniste and Merrill, 1986) or inhibit (Knobler et al., 1988) differentiation of oligodendrocytes depending on the microenvironment. Therefore, corticosteroids may have promoted remyelination in vivo by inhibiting a pathogenic component from the microenvironment, thereby allowing the normal physiological response to predominate. Of interest, SCH94.03 has also been shownin vivo to have local immunosuppressive properties because treatment with the antibody successfully prevents further relapses of experimental autoimmune encephalomyelitis (Miller et al., 1997) and suppresses CNS inflammation after Theiler’s virus-induced demyelination (Miller et al., 1996). This effect appears to be primarily a T-cell rather than macrophage phenomenon, which may explain the dissociation of mechanism of action between methylprednisolone and SCH94.03, as supported by the results (Fig. 6).
Second, treatment may have promoted remyelination by actively stimulating myelination in vivo. Corticosteroids, either exogenously administered or endogenously synthesized by glial cells (Jung-Testas et al., 1989), have been shown to promote gene transcription (Gronemeyer, 1992), in particular on rat glial cultures (Kumar et al., 1989). Glucocorticoids are also co-mitogens for Schwann cells (Neuberger et al., 1994). The mAb SCH94.03 may also have had a direct stimulatory effect on oligodendrocytes (Asakura et al., 1996a). mAb SCH94.03 labels a surface antigen on a subpopulation of mature murine oligodendrocytes raising the possibility that it promotes remyelination by binding directly to a surface receptor (Asakura et al., 1996). The mAb SCH94.03 also localizes to the lesions afterin vivo administration and binds to glia, myelin, macrophages, and some axons (Hunter et al., 1997). Other antibodies that directly stimulate oligodendrocyte differentiation include the O4 mAb (Bansal et al., 1988) and an mAb against a reovirus receptor expressed by oligodendrocytes (Cohen et al., 1991). Therefore, methylprednisone and mAb SCH94.03 may have promoted remyelination by stimulating directly remyelination by oligodendrocytes.
At the inception of these experiments we predicted that those therapeutic strategies that would have enhanced remyelination also would have resulted in increased [H3]thymidine uptake in the lesion. This was a logical conclusion based on elegant previous studies demonstrating that proliferation of progenitor glial cells is necessary before remyelination (Ludwin, 1979; Armstrong et al., 1990; Prayoonwiwat and Rodriguez, 1993; Carrol and Jennings, 1994). However, no statistical differences were observed in the number of proliferating cells in the lesions at 3 weeks after the various strategies to promote remyelination. We cannot exclude the possibility that a change in the number of proliferating cells between the various groups would have been seen if examined before 3 weeks. Although we did not do double-labeling experiments to identify the phenotype of these cells, previous experiments in other models of demyelination indicate that the majority of the proliferating cells are glial (Armstrong et al., 1990; Prayoonwiwat and Rodriguez, 1993). The results of our experiments indicate that the stimuli for inducing proliferation, a likely prerequisite for remyelination, are independent of the treatment paradigms we used. The result suggests that once these cells have proliferated, the approach used here allows for more rapid and efficient myelin synthesis.
Treatment with mAb SCH94.03 increased statistically the number of remyelinated axons per lysolecithin lesion area at 3 weeks, whereas polyclonal Ig and anti-SCH Ig did not enhance remyelination using the same dose and duration of treatment as in our TMEV studies (Rodriguez and Lennon, 1990; van Engelen et al., 1995). These results are intriguing and clearly indicate that with increasing specificity of the Ig preparation (from polyclonal Ig, polyclonal anti-SCH Ig, to mAb SCH94.03) remyelination also increased (Table 2). This raises the possibility that remyelination-promoting antibodies may be present in normal serum but at a low concentration. The mAb SCH94.03 has the phenotype of naturally occurring or physiological autoantibodies, which are found in the serum of normal humans and mice, are polyreactive with a wide range of antigens (Asakura et al., 1996b), and are encoded by germ line Ig light- and heavy-chain genes without definitive somatic mutations (Miller et al., 1995). The enhancement of remyelination in the lysolecithin model implies that certain natural autoantibodies may participate in a beneficial physiological response to CNS injury, even in nonimmune demyelination.
Footnotes
This work was supported by the Dutch Society for Support of Research on Multiple Sclerosis (Grant 92-115m to B.v.E.) and by National Institutes of Health Grant NS24180. We also appreciate the generous financial support of the Myelin Project.
K.D.P. and B.G.M.v.E. contributed equally to this manuscript.
Correspondence should be addressed to Dr. Moses Rodriguez, Department of Neurology, Mayo Clinic and Mayo Foundation, Rochester, MN 55905.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}