Article Text

Abstract
Introduction: Mutations in the voltage-gated sodium channel SCN1A gene are the main genetic cause of Dravet syndrome (previously called severe myoclonic epilepsy of infancy or SMEI).
Objective: To characterise in more detail the mutation spectrum associated with Dravet syndrome.
Methods: A large series of 333 patients was screened using both direct sequencing and multiplex ligation-dependent probe amplification (MLPA). Non-coding regions of the gene that are usually not investigated were also screened.
Results: SCN1A point mutations were identified in 228 patients, 161 of which had not been previously reported. Missense mutations, either (1) altering a highly conserved amino acid of the protein, (2) transforming this conserved residue into a chemically dissimilar amino acid and/or (3) belonging to ion-transport sequences, were the most common mutation type. MLPA analysis of the 105 patients without point mutation detected a heterozygous microrearrangement of SCN1A in 14 additional patients; 8 were private, partial deletions and six corresponded to whole gene deletions, 0.15–2.9 Mb in size, deleting nearby genes. Finally, mutations in exon 5N and in untranslated regions of the SCN1A gene that were conserved during evolution were excluded in the remaining negative patients.
Conclusion: These findings widely expand the SCN1A mutation spectrum identified and highlight the importance of screening the coding regions with both direct sequencing and a quantitative method. This mutation spectrum, including whole gene deletions, argues in favour of haploinsufficiency as the main mechanism responsible for Dravet syndrome.
Statistics from Altmetric.com
Severe myoclonic epilepsy of infancy (SMEI; OMIM 607208) or Dravet syndrome is an intractable epileptic encephalopathy occurring in the first year of life in a previously normal infant. Patients present with recurrent and prolonged seizures, usually febrile, hemiclonic or generalised tonic clonic, resulting commonly in status epilepticus.1 Patients later develop other seizure types including atypical absence and partial seizures concomitant with developmental delay, ataxia and myoclonic jerks.1 2 Cognitive outcome is poor, with a high risk of mental retardation. Dravet syndrome is usually sporadic but familial cases have been occasionally reported.3
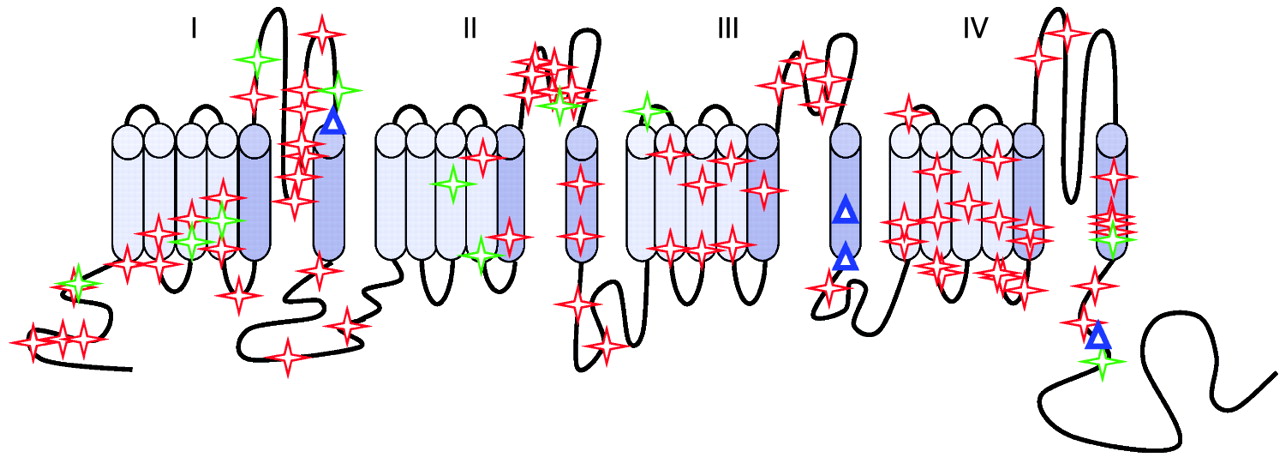
Heterozygous mutations in SCN1A, the gene encoding the voltage-gated neuronal sodium channel alpha 1 subunit (Nav1.1), are a major cause of Dravet syndrome.4 The Nav1.1 protein belongs to a protein family that includes 10 members in humans, which are involved in the genesis of action potentials and rapid membrane depolarisation. These channels contain four transmembrane domains (I–IV), each with six membrane-spanning segments (S1–S6). All types of mutations in SCN1A are observed in patients with SMEI, including missense mutations and all types of truncating mutations.5 Recently, large intragenic deletions, undetected by standard techniques, have also been described.6–10 This large mutation spectrum contrasts with that of generalised epilepsy with febrile seizures plus (GEFS+; OMIM 604233), an autosomal dominant condition also associating with febrile and afebrile seizures but with a usually benign outcome, in which only missense mutations in SCN1A are found.11 Loss of function of the mutated allele is probably responsible for SMEI, as confirmed by the recent development of knock-out and knock-in mouse models,12 13 whereas another mechanism could lead to GEFS+.
Several isoforms of SCN1A have been reported although the isoform(s) relevant for the physiopathology of Dravet syndrome remain(s) unknown: three different donor sites of intron 11 lead to alternative spliced isoforms containing short, intermediate or long versions of exon 11, effect of which is to modulate the length of the intracellular loop between segments IS6 and IIS1.14 Furthermore, an exon 5N has been described, highly homologous to the exon 5 normally present in the adult isoform (exon 5A).15 Replacement of exon 5A by exon 5N has been suggested to be developmentally regulated and to occur at a fetal stage, as previously described for SCN8A.16 Recently, novel 5′ untranslated region (UTR) exons of SCN1A have been described; some of these 5′UTR exons and additional intronic regions have been shown to be highly conserved during evolution.17
The aim of this study was to screen a large series of patients with Dravet syndrome using combined approaches allowing both the detection of point mutations and exonic rearrangements. In addition, we analysed exon 5N and eight of the regions of the SCN1A gene conserved during evolution, to determine whether additional mutations could be found in these regions.
METHODS
This study was approved by the ethics committee (CCPPRB of Pitié-Salpêtrière Hospital, Paris, no 69-03, 25/9/2003). Informed written consent was obtained from the patients’ parents before blood sampling.
Patients
In total, 333 probands diagnosed with Dravet syndrome were referred to our diagnostic laboratory for genetic testing of SCN1A in a 5-year period between April 2003 and April 2008. Most samples were received from specialised child neurology centres throughout France although a few samples were occasionally received from other countries (Slovenia, Germany, and Chile).
The referral physician completed a detailed clinical questionnaire for every patient. Clinical reports were also collected for most patients to assess the clinical history of the disease. All clinical files and questionnaires were examined by the same neuropaediatrician and neurologist (RN and IA) for consistent classification of the patients. Core features required for classification as classic Dravet syndrome were defined as: normal cognitive and motor development previous to seizure onset, onset of the seizures before 1 year of age, seizures mainly triggered by fever, long-lasting seizures (>15 min, that might evolve to status epilepticus), later occurrence of various seizures types (febrile and afebrile) and later cognitive regression. The presence of myoclonic jerks and/or ataxia was considered as a highly characteristic feature reinforcing the diagnosis but their absence did not exclude the clinical diagnosis of Dravet syndrome as both were previously shown to be inconsistent in patients.
In total, 271 patients had clinical features that conformed to this description. The remaining 62 patients were not considered as having typical Dravet syndrome: 6 patients had a clinical history highly suggestive of Dravet syndrome but were still too young (age<12, 13 and 15 months respectively) to fulfil all the clinical criteria, 6 patients had an onset after 1 year of age (12.5–15 months), 1 patient had a very early onset (10 days), 15 patients were reported as having only GTC although they fulfilled all other criteria, and 34 patients diagnosed with Dravet syndrome by their local neuropaediatrician were not classified due to absent or incomplete clinical information. Patients were first screened using direct sequencing of SCN1A coding sequence and patients without point mutation were subsequently analysed for exon deletion using MLPA.
Analysis of the SCN1A coding sequence by direct sequencing
The 30 specific primer pairs amplifying the 26 exons of the SCN1A gene (transcript reference AB093548) have been described previously.3 18 Primers specific to exon 5N and to eight regions of SCN1A, located in the 5′UTR or in introns 11 and 20 and highly conserved in orthologues and paralogues of SCN1A,17 were designed to amplify the corresponding regions (supplementary table A online). Sequence products were run on an automated sequencer (ABI 3730; Applied Biosystems, Foster City, California, USA) and data were analysed with Seqscape V.2.5 software (Applied Biosystems). Mutations found in patients were investigated in available parents using direct sequencing of the corresponding exon. When the mutation was absent from both parents, parental testing was performed using microsatellite markers at the SCN1A locus to ensure that the mutation occurred de novo. In addition, 180 European controls (healthy spouses of patients with other neurological diseases) were included to test new variants of the SCN1A gene.
Multiplex ligation-dependent probe amplification
Multiplex ligation-dependent probe amplification (MLPA) was used to search for rearrangements in SCN1A,6 using a commercial kit (MLPA P137 Kit; MRC-Holland, Amsterdam, The Netherlands). MLPA reactions were carried out according to the manufacturer’s instructions. Electrophoresis of PCR products was performed using an automated sequencer (ABI 3730; Applied Biosystems) and MLPA data were analysed using GeneMapper V.3.5 software (Applied Biosystems). Relative ratios were calculated using the formula r = mean (peak areapatient/control areapatient)/(peak areacontrol/control areacontrol).
Characterisation of SCN1A deletions breakpoints using high-density single-nucleotide polymorphism arrays
Patients with large SCN1A deletions were further analysed using Illumina 370CNV-Duo genotyping BeadChip arrays (370 K). The genotyping reaction steps were performed according to the manufacturer’s specifications (Infinium II; Illumina, San Diego, California, USA). Image analysis and automated genotype calling was performed using Beadstudio V.3.1 (Illumina). Breakpoints of the deletions were defined as the first and last single-nucleotide polymorphism (SNP) that were homozygous and presented a decreased log ratio (in the order of −0.5) in the region of the deletion.
Bioinformatics analyses of SCN1A mutations
Mutation interpretation and amino acid conservation in orthologues was assessed using Alamut V.1.31 software (Interactive Biosoftware, Rouen, France; http://www.interactivebiosoftware.com/). For missense mutations, the Grantham chemical distance between amino acids19 provided by the Alamut software was used to test whether the replacements between the residues are important with respect to a range of physiochemical properties (volume, charge and composition of the side chain).
RESULTS
In total, 242 patients (73%) with SCN1A mutations (supplementary table B online) or deletions (table 1) were identified. A schematic representation of the localisation and nature of the mutations on the SCN1A gene is shown in fig 1.
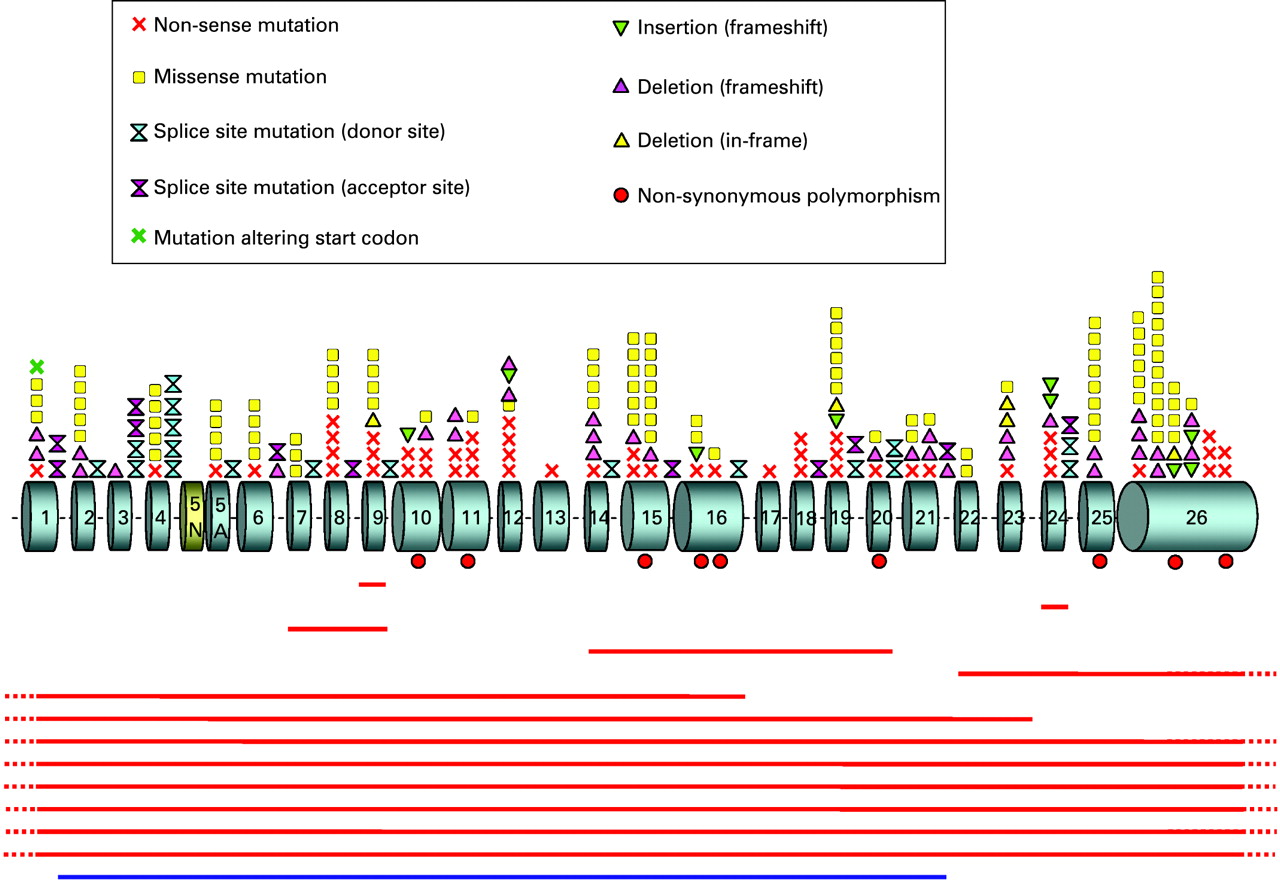
Point mutations detected by direct sequencing
Direct sequencing detected 200 different heterozygous mutations, 161 of which were novel, in 228 patients (supplementary table B online). Point mutations leading theoretically to missense amino acids substitutions were the most common mutation type, encountered in 42% of the patients (96/228). Other mutation types introducing premature termination codon (PTC) were also common: nonsense mutations (23%, 52/228), mutations altering splice sites (13%, 29/228), and small deletions or insertions leading to a frameshift (19%, 44/228). Finally, we identified five in-frame deletions (2%, 5/228), one mutation altering the adenine of the ATG initiation codon, and one intronic substitution located 10 bp upstream from exon 16, which was classified as a mutation on the basis that it occurred de novo in the patient although it has no predicted effect on SCN1A splicing (0.5% each, 1/228).
Interestingly, we found four patients harbouring two SCN1A variants with possibly deleterious effect: one carried the c.2816A→C/p.His939Pro and c.5364C→A/p.Asn1788Lys variants; the second had the c.3235G→A/p.Val1079Ile variant, inherited from his asymptomatic father, associated with the de novo c.2504_2508delTTGAC mutation; the third carried the c.4723C→T/p.Arg1575Cys variant, inherited from his asymptomatic mother, and the de novo c.1804G→T/p.Glu602X mutation; and the fourth had the c.3325C→A/p.Pro1109Thr, inherited from his father, and the de novo c.4133delA mutation. We hypothesised that p.Val1079Ile, p.Pro1109Thr and p.Arg1575Cys were novel rare non-synonymous polymorphisms, but we could not define which of the p.His939Pro and p.Asn1788Lys variants constituted the causative mutation, as the parents of this patient were unavailable for genetic analyses. Therefore, both were considered as potential missense mutations.
Most missense mutations alter a highly conserved amino acid in the protein (96%, 92/96 different missense mutations; supplementary table C online). Furthermore, the amino acid substitutions involve residues that are chemically dissimilar as assessed by the Grantham distance formula;19 71% of the missense mutation show a distance >70, corresponding to dissimilar or very dissimilar changes (68/96); 77% alter a residue belonging to ion transport sequences (ie, sequences sharing significant homology to other ion channels and transporters; 74/96) or both (58%, 56/96). However, the missense mutations were located throughout the Nav1.1 channel, with no preferential location in transmembrane, extracellular or intracellular domains (fig 2). Interestingly, all the mutations found in exon 11 are located in the portion shared by all three alternatively spliced isoforms of this exon (ie, in the shortest isoform) and no variant or mutation specific to the long isoforms have been described to date.
For 149 patients with point mutation, both parents were available for genetic analyses. Direct sequencing failed to detect the mutation in either parent, in 133 cases out of 149 (89%), indicating that the mutation occurred de novo in these patients. For the 15 patients who inherited the mutation, it was inherited from the mother in 5 cases and from the father in 10. Two siblings (brother and sister) had the same mutation although it was undetectable in their parents. Interestingly, we found 9 patients with novel recurrent mutations (ie, an identical mutation in >1 patient) and 67 patients had mutations that have previously been reported by other authors (supplementary table B online). However, most mutations occurred de novo, indicating that they are repetitively generated through specific mechanisms. Further analysis of patients with inherited mutations is ongoing (Depienne et al, in preparation).
In total, 40 polymorphisms, 24 of which are novel, were identified in SCN1A (supplementary table D online), including polymorphisms detected in the relatives of asymptomatic patients and in controls. The eight non-synonymous variants (p.Arg542Gln, p.Arg604His, p.Ala924Thr, p.Val1079Ile, p.Pro1109Thr, p.Glu1308Asp, p.Arg1575Cys, and p.Arg1928Gly) were present in an asymptomatic parent, suggesting that they are benign variants; however, they were not found in 100 Caucasian controls, indicating that they are either rare or possibly specific to populations with a particular geographical origin. Surprisingly, all these variants, with the exception of p.Ala924Thr, also affect highly conserved amino acids of the proteins, but compared with the missense mutations found in patients, they mainly substitute chemically similar amino acids (33% (3/9) have a distance >70 arbitrary units (AU), corresponding to dissimilar or very dissimilar changes, compared with 74% for missense mutation) and, with the exception of p.Arg1575Cys, none transforms a highly conserved residue belonging to ion-transport sequences in a chemically dissimilar residue.
Microdeletions detected with MLPA and characterisation using microarrays
MLPA analysis of the 105 patients without point mutations found in 14 patients a heterozygous rearrangement in SCN1A, encompassing a variety of lengths from a single exon to the whole gene (13%, 14/105; table 1). Deletion of the whole gene was the most common abnormality, which was found in six patients (43%, 6/14), one of whom has an affected brother with the same deletion. The eight other partial rearrangements, including seven deletions (exon 9, exon 24, exons 1–16, exons 1–23, exons 14–20, exons 22–26, and exons 7–9 found in monozygous twins) and one duplication (exons 2–21) were all private and novel. Both parents of the three patients with deletions of exons 22–26, 1–16, and 7–9, respectively, were available for analysis, and this showed that all three deletions occurred de novo. Parents of the brothers with the whole gene deletion were unavailable although the presence of the same deletion in them indicates an inherited rearrangement.
To characterise the size and breakpoints of deletions encompassing the first and/or last exon of SCN1A, we analysed the nine corresponding patients using high-density SNP arrays (370 K; Illumina), and found that the eight detectable deletions have different breakpoints and size, (range 0.15 to 2.9 Mb), with the exception of two deletions (patients 3 and 4) with similar size and close breakpoints (figs 3,4). The nearby genes comprised in the deletion were therefore different between patients. The remaining deletion, covering exons 22–26, was the smallest and could not be detected with this method. The values obtained for the SNP in the region suggest that it is limited to the SCN1A gene. These results show that most genomic deletions encompassing SCN1A are generated through different molecular mechanisms.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Screening of additional regions of SCN1A in negative patients
Tate et al15 have described a highly conserved alternative exon (5N), located 95 bp upstream of the exon 5 present in the adult isoform (exon 5A), which introduces three differences in the 30 encoded amino acids (Tyr202Phe, Asp208Asn and Val212Phe) when incorporated instead of exon 5A.15 The functional consequence of this substitution is to date unknown. However, the switch between these two isoforms is thought to be developmentally regulated, as has been shown for SCN8A, with transcripts containing exon 5N predominating in fetal and neonatal brain, and transcripts containing exon 5A being the preferential isoform expressed in adult brain. We identified several different types of mutation in exon 5A in our patients with Dravet syndrome, confirming previous studies.5 To determine whether mutations or variants in exon 5N can be identified in patients with Dravet syndrome, we directly sequenced this exon in the remaining negative patients. No deleterious change was detected in exon 5N, indicating that this exon and the corresponding isoform are probably not involved in the physiopathology of the disease.
Recently, Martin et al17 described seven novel non-coding exons of SCN1A (1a to 1g, corresponding to the 5′ UTRs). Furthermore, comparison with paralogous (SCN1A vs SCN2A and SCN3A) and orthologous (human vs mouse) sequences revealed 11 conserved non-coding sequences in SCN1A (in the 5′ UTR, intron 10 and intron 20), at least two of which could influence the transcription of the gene.17 To determine whether variants or mutations in these regions can cause Dravet syndrome, we screened the eight most conserved regions in our negative patients. Eight different variants (two in the 5′UTRs, one in intron 11 and five in intron 20) were identified in seven patients (supplementary table E online). Analysis of the parents of three patients revealed that the c.1-52589T→C (5′UTR), c.2043+233C→T (intron 11), c.4003-661A→G, c.4002+2052T→C and c.4003-672_668delATTAT (intron 20) mutations were inherited from an asymptomatic parent. In addition, two variants (c.4002+2052T→C and c.4003-661A→G) were also identified in a Caucasian control population (supplementary table E online). These results suggest that none of the variants detected in the 5′UTR and intron 20 are involved in Dravet syndrome, and that mutations outside the coding region of SCN1A are not a common cause of Dravet syndrome for patients without mutations in the coding region.
In conclusion, this analysis found that 242 of 333 (73%) patients carried a mutation: 228 point mutations and 14 microrearrangements (8 partial SCN1A deletions, 3 of which were partial deletions extending outside SCN1A and 6 whole gene deletions).
DISCUSSION
This study reports the wide mutation spectrum of SCN1A associated with Dravet syndrome, including both point mutations and rearrangements, in the largest series of patients reported to date. In addition, we screened the conserved non-coding regions of the gene and excluded causative mutations in these regions in patients negative for mutations in the coding sequence.
Our results confirm that all types of DNA alterations in the SCN1A gene are present in patients with Dravet syndrome: missense, nonsense, and splice-site mutations, small deletions and insertions, and rare large-scale deletions. Mutations in our patients were found all along the SCN1A gene. Missense mutations were the most common mutation type identified in our patient group (42%), in contrast to the initial report of Claes et al, which reported only truncating mutations.4 The main remaining mutation types (nonsense, splice-site mutations, frameshift) introduce PTC into the mRNA, which are probably recognised and degraded via the nonsense-mediated mRNA decay surveillance system of the cell, with the exception of truncating mutations located in exon 26.20 This hypothesis is compatible with the effects of the other mutation types detected including alteration of the initiation codon and the microdeletions that delete essential regions of the proteins, introduce PTC, or delete the whole gene in the heterozygous state. Consequently, the preferential mechanism suggested by this mutation spectrum would be haploinsufficiency ie, total loss of function of the mutated allele.
In spite of many functional studies, it remains unclear how missense mutations can cause a clinical phenotype indistinguishable from that of truncating mutations or whole-gene deletion. Kanai et al suggested a preferential location of the missense mutations in the S5 and S6 segments and the S5–S6 intracellular boucle, which together form the “pore” of the Nav1.1 channel.21 The systematic classification of missense mutations using Alamut software revealed that most missense mutations found in our study affect highly conserved amino acids located in ion-transport sequences and result in chemically dissimilar changes in amino acid classes. However, these mutations were not preferentially located in S5–S6 segments, in contrast to the previous report.21
In addition to mutations detected by standard techniques, we identified 13 patients with partial (n = 7) or complete (n = 6) gene deletions and one patient with a partial gene duplication. This is the largest series of patients with rearrangements in SCN1A, and the first duplication reported to date. It raises the number of partial rearrangements in SCN1A to 14, including the 6 intragenic deletions previously reported.6 9 10 Whole-gene deletion was the main event detected, but analysis of the patients with this abnormality using high-density SNP arrays revealed that, with the exception of two patients who shared a deletion similar in size, all other deletions were private rearrangements that also delete nearby genes in the 2q24 region, including in particular other genes encoding voltage-gated sodium channels (SCN7A and SCN9A). In the three patients with partial deletion for whom the parents were available, the deletion was confirmed to have arisen de novo. The presence of two brothers with the same complete gene deletion showed that occasionally, rearrangements can also be inherited from an asymptomatic parent, as already described for point mutations. The mechanisms involved in these inherited mutations will be investigated in detail elsewhere (Depienne et al, in preparation).
Strikingly, the patients with these large deletions had similar clinical features compared with those with point mutations, and none presented with additional features. These results suggest that the haploinsufficiency of these voltage-gated sodium channels genes, in which missense mutations have been occasionally reported in patients with various epileptic phenotypes, including benign familial neonatal–infantile seizures,22 23 does not worsen the clinical features of patients with SMEI. On the other hand, four patients in previous studies were reported to have features additional to SMEI: one patient had autism, precocious puberty and palatoschisis;7 the second had features of a neurodegenerative disorder including acquired microcephaly, psychomotor and visual regression, very severe photosensitive epilepsy, athetosis of the four limbs, and hepatomegaly;8 the third had dysmorphic features, growth failure, hypotonia, cardiac defects and limb abnormalities;24 and the fourth presented with muscular hypotonia, dysmorphy, cleft palate, high anal atresia, atrial septal defect and syndactyly.25 However, most of these clinical features, which differ between patients, are unlikely to be due only to haploinsufficiency of genes located in these regions, as at least one patient with “pure” Dravet syndrome had a deletion encompassing these genes (fig 3), with the exception of the last patient who had the largest distal deletion.25 Nevertheless, in the remaining cases, the deletion could reveal mutations or variants located in one of these genes on the undeleted allele, which could contribute to the clinical picture. The additional features could also be due to other alterations unrelated to the 2q24 (SCN1A) deletion in the genome of the patients. However, several very large deletions encompassing more than the 2q24.3 chromosomal band have been reported (for review see Davidsson et al25). In contrast to patients identified and reviewed in this study, patients with these very large deletions could have consistent dysmorphic features including ear abnormalities, microcephaly, micrognathia and brachysyndactyly.25
The proportion of patients with SCN1A mutations in our series is 73% (242/333). This proportion has been highly variable in previous studies, ranging from 33%18 26 to 80–100%.4 5 27 28 These discrepancies may be due to the sizes of the series and the use of different clinical criteria to define Dravet syndrome. Supporting this hypothesis, the proportion of positive patients is higher in SMEI using strict criteria than in epileptic syndromes closely related to SMEI including ICEGTC (intractable childhood epilepsy with generalised tonic–clonic seizures) or SMEB (borderline SMEI).27–29 In our series, we also had to take the various clinical presentations of the patients into account (see Methods). Considering only the patients with typical Dravet syndrome (n = 271), the proportion of patients with SCN1A mutation reaches 78% (212/271). These results confirm that approximately 20% of patients presenting with classic features of Dravet syndrome do not have mutations in SCN1A, even when microrearrangements have been excluded. These results validate the mutation frequencies recently reported in the literature.10 29 In addition, recent studies have established that rare pathogenic mutations in SCN1A, some of which are de novo, can also be identified in other infantile epileptic encephalopathies such as cryptogenic generalised epilepsies, cryptogenic focal epilepsies or infantile spasms.26 29 30 This extension of the clinical spectrum related to SCN1A has questioned the initial concept of SMEI, because neither clinical nor genetic criteria are sufficient to delimit accurately the various syndromes. Interestingly, we report two mutations, p.Arg393Cys and p.Arg1596Cys (previously identified in patients with cryptogenic focal epilepsy29) in patients with typical SMEI. This suggests that the variation in clinical presentation is not intrinsic to the mutations themselves but rather to their interaction with other yet unidentified genetic or environmental factors. We also report the p.Arg1575Cys variant, recently identified in a patient with Rasmussen encephalitis,31 in a patient who had another de novo p.Glu602X mutation and in his asymptomatic mother. This variant has been shown to increase persistent sodium current compared with the normal channel in in vitro studies.31 Our results confirm that this rare variant is not associated with Dravet syndrome. Neither this variant nor another SCN1A variant was found in three patients with Rasmussen syndrome (C Depienne & R Nabbout, personal communication). The study of additional patients with this variant is needed to confirm whether it is a risk factor for Rasmussen syndrome.
As SCN1A is the main gene involved in Dravet syndrome, we hypothesised that other mutation types located in this gene but missed using both direct sequencing and MLPA, could be identified in negative patients. We therefore screened exon 5N, which is suspected of involvement in early fetal stages of the development, and highly conserved non-coding regions of the gene, which could be important for normal gene expression and regulation. However, this screening did not reveal any additional mutation, indicating that mutations outside the coding region of SCN1A are uncommon and suggesting that Dravet syndrome is genetically heterogeneous. Further genetic studies are needed to determine the causative mutations and genes involved in the remaining SCN1A-negative patients.
Acknowledgments
We thank the families for participating to this study and to the clinicians who referred their patients: Professors O Dulac, M Baulac and V Des Portes and Drs A Afenjar, C Altuzzara, F Audic-Gérard, N Bahi-Buisson, C Barnerias, M-A Barthez-Carpentier, P Berquin, N Bednarek, P Bèze-Beyrie, T Billette de Villemeur, O Boesflug-Tanguy, D Bonneau, S Briault, C Brière, M Bru, C Cances, P Castelnau, I Caubel, S Chabrier, B Chabrol, D Chaigne, N Chassaing, M-C Chevalier, E Cheuret, M Chipaux, C Chiron, M-A Cournelle, C De Barace, B Delobel, A De Saint-Martin, I Desguerre, D Doummar, C Dravet, V Drouin-Garraud, S Dupont, A Dusser, C Espil-Taris, V Flurin, J Gaudelus, F Giulano, D Graber, A Guet, L Guyant-Marechal, E Hirsch, M Husson, C Ioos, L Issa-Brunet, P Jonveaux, P Jouvencel, A Kaminska, M Lackmy Port-Lis, E Lagrue, L Lambert, P Landrieu, A Le Bihannic, S Lebon, B Leheup, M-N Loiseau-Corvez, M-O Livet, J Mancini, S Marignier, J Motte, S Napuri, V Navarro, E Panagiot, J-M Pédespan, B M Perkovic, S Peudenier, J-M Pinard, G Plessis, I Py, C Ringenbach-Rivier, F Rivier, A Roubertie, S Rivera, P Sabouraud, P Sanyas, L Rios Pohl, L Servais, S Sukno, D Taussig, C Thauvin, D Ville, N Villeneuve, Y Wardi, C Zix. We thank Genethon for the DNA extraction and banking of a part of patients reported in this series. This work was financially supported by the Programme Hospitalier de Recherche Clinique AP-HP (n°P020910/AOR02085).
conflict of interest and no financial interest in this study.
REFERENCES
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online Table A
- Data supplement 2 - Online Table B
- Data supplement 3 - Online Table C
- Data supplement 4 - Online Table D
- Data supplement 5 - Online Table E
Footnotes
Competing interests: None.
▸ Supplementary tables are published online only at http://jmg.bmj.com/content/vol46/issue3