Article Text

Abstract
Background Expanded GGGGCC hexanucleotide repeats in the promoter of the C9ORF72 gene have recently been identified in frontotemporal dementia (FTD), Amyotrophic Lateral Sclerosis (ALS) and ALS-FTD and appear as the most common genetic cause of familial (FALS) and sporadic (SALS) forms of ALS.
Methods We searched for the C9ORF72 repeat expansion in 950 French ALS patients (225 FALS and 725 SALS) and 580 control subjects and performed genotype-phenotype correlations.
Results The repeat expansion was present in 46% of FALS, 8% of SALS and 0% of controls. Phenotype comparisons were made between FALS patients with expanded C9ORF72 repeats and patients carrying another ALS-related gene (SOD1, TARDBP, FUS) or a yet unidentified genetic defect. SALS patients with and without C9ORF72 repeat expansions were also compared. The C9ORF72 group presented more frequent bulbar onset both in FALS (p<0.0001 vs SOD1, p=0.002 vs TARDBP, p=0.011 vs FUS, p=0.0153 vs other FALS) and SALS (p=0.047). FALS patients with C9ORF72 expansions had more frequent association with FTD than the other FALS patients (p<0.0001 vs SOD1, p=0.04 vs TARDBP, p=0.004 vs FUS, p=0.03 vs other FALS). C9ORF72-linked FALS patients presented an older age of onset than SOD1 (p=0.0139) or FUS mutation (p<0.0001) carriers. Disease duration was shorter for C9ORF72 expansion carriers than for SOD1 (p<0.0001) and TARDBP (p=0.0242) carriers, other FALS (p<0.0001) and C9ORF72-negative SALS (p=0.0006).
Conclusions Our results confirm the major role of expanded repeats in C9ORF72 as causative for ALS and provide evidence for specific phenotypic aspects compared to patients with other ALS-related genes.
- Motor neuron disease
- familial ALS
- Genetic analysis
- repeat primed PCR
- GGGGCC repeat
- genetics
- Parkinsons disease
- molecular genetics
- genetic screening/counselling
- epilepsy and seizures
Statistics from Altmetric.com
- Motor neuron disease
- familial ALS
- Genetic analysis
- repeat primed PCR
- GGGGCC repeat
- genetics
- Parkinsons disease
- molecular genetics
- genetic screening/counselling
- epilepsy and seizures
Introduction
Amyotrophic Lateral Sclerosis (ALS) is a fatal adult onset motor neuron disease with familial transmission in 6%–10% of the cases. Mutations in SOD1 encoding the cooper/zinc superoxide dismutase, TARDBP (TAR DNA-binding protein 43) and FUS (fused in sarcoma) occur in 20%–30% of familial forms of the disease (FALS).1 Rare cases of ALS are also linked to mutations in ANG, VAPB, DAO, OPTN, VCP and UBQLN2.1–5 As ANG encodes a pancreatic ribonuclease with regulatory functions on ribosomal RNA transcription and TARDBP and FUS encode proteins with putative similar DNA/RNA-binding functions, defects in RNA processing pathways are an appealing hypothesis for ALS disease. The recent discovery of expanded hexanucleotide repeats in a non coding region of C9ORF72 in ALS, frontotemporal dementia (FTD), and ALS-FTD6–8 supports this view since these repeats seem to lead to the downregulation of the expression an alternatively spliced C9ORF72 transcript and to the formation of nuclear RNA foci. In this study we analysed 950 French ALS patients to evaluate the frequency of the repeat expansion in C9ORF72 in FALS and SALS, and compared clinical phenotypes shown by these patients to patients with mutations in other ALS related genes.
Methods
Patients
FALS included 225 index cases of unrelated families with probable or definite ALS,9 131 males and 94 females (M:F ratio=1.4:1) with mean age of onset at 55 years (SEM 1, median 55 years, range 21–85 years,) and mean disease duration of 51 months (SEM 4, median 33 months, range 3–336 months, including 32 censored data). SALS included 725 patients, 420 males and 305 females (M:F ratio=1.4:1), with mean age of onset at 56 years (SEM 1, median 58 years, range 21–87 years) and mean disease duration of 72 months (SEM 4, median 48 months, range 1–354 months, including 90 censored data). Data were censored at the last date of the patients visit. Control samples were age-matched Caucasian individuals of French background (n=580). Most DNA samples were collected over the past 15 years at the ALS National Referal Center of Pitié-Salpêtrière Hospital (Paris). During the same period, the number of ALS patients followed by the center was 7784. Some families were collected by other French ALS Centers belonging to the French ALS study group. Criterion for family inclusion was that at least two members were affected. In FALS the disease was transmitted in a dominant (n=164) or putatively recessive (all affected members belonged to a single sibling with none of the parent affected, n=47) manner. In 14 remaining families, the ALS patients were distant relatives (more than 2nd degree relatives). All participants signed a consent form allowing performing research. Protocols were approved by the Medical Research Ethics Committees of the ‘Comité d’Ethique de la Pitié-Salpêtrière’ and ‘Assistance Publique-Hôpitaux de Paris'.
Genetic analysis
All FALS have previously been screened for SOD1, ANG, TARDBP, FUS, DAO and OPTN.10–12 FALS without male-to-male transmission (n=130) had also been analysed for the X linked UBQLN2 gene.13 Patients with mutation in SOD1 (n=26), ANG (n=1), TARDBP (n=8), FUS (n=13), DAO (n=1), OPTN (n=2), UBQLN2 (n=1) and with no previously identified genetic defect (n=173) were included in the C9ORF72 repeat analysis. For each patient, sequencing of ALS related genes and determination of C9ORF72 repeat length were performed on the same DNA sample.
The analysis of the C9ORF72 repeat was performed by a repeat-primed PCR amplification as previously described.6 This analysis was completed by a classical fluorescent fragment-length analysis allowing the detection of non-expanded C9ORF72 alleles.6 Both analyses were repeated twice for each patient sample to ensure reproducibility of the results, determine whether the repeat expansion was present at the heterozygous or homozygous state and because repeat primed assay efficiency highly depends on DNA concentration and quality. DNA of some homozygote individuals was sequenced to precisely correlate the number of base pairs in the fluorescent assay with the number of GGGGCC repeats. For clinical comparison, only patients with GGGGCC repeat numbers greater than 50 were included in the C9ORF72 group. Although no biological data is available to support that 50 repeats is a suitable cut-off to determine pathogenicity, this 50 repeat cut-off corresponds to the detection limit of the method we used.
Clinical analysis
Clinical data could be partially recovered for up to 334 relatives belonging to the 225 analysed families and 725 SALS cases. The exact number of patients included in each statistical analysis depended on the availability of the corresponding data and is summarised in tables 1 and 2. Clinical parameters including gender ratio (n=334 FALS, 719 SALS), age of onset (n=290 FALS, 512 SALS), site of onset (n=283 FALS, 703 SALS), disease duration (n=264 FALS, 512 SALS)10 and presence of FTD behavioural variant (n=162 FALS)14 were compared in five FALS groups represented by patients with long C9ORF72 repeat, mutations in SOD1, TARDBP, FUS or other FALS with still unidentified mutations (table 1). In addition, 2 groups of SALS patients either positive or negative for expanded C9ORF72 repeats were included. Disease duration was also compared between C9ORF72 patients with bulbar (n=42) or spinal (n=70) onset and with (n=25) or without (n=28) FTD. FALS were arbitrarily separated according to age of onset (≤40, 41–50, 51–60, 61–70, ≥71) to study the frequency of ALS-related genes in each subgroup. Two consecutive subgroups were pooled when their distributions were similar.
Clinical comparison between groups of patients with a mutation in SOD1, TARDBP, FUS, hexanucleotide repeats in C9ORF72 and patients with no identified genetic defect (other FALS)
Clinical comparison between groups of SALS patients with or without expanded repeats in C9ORF72
Statistical analysis
Statistical analyses were performed as previously described.10 Briefly, the Cox proportional hazards regression model was used to compare age at onset and disease duration between the five groups of FALS patients. If the difference was significant (p<0.05), a log rank analysis compared groups by pairs. Proportions of patients classified according to gender, site of onset or presence of FTD were compared by pairs using Fisher's exact tests. Proportions of C9ORF72-positive subgroup of patients (n=70) with available cognitive status data (FTD or pure ALS) classified according to site of onset (bulbar or spinal) were also compared using Fisher's exact test. Statistical analyses were performed using the SPSS V.11.0 data analysis software (SPSS Inc.).
Results
In controls, the mean number of C9ORF72 repeat expansions was of 4 (range = 2–23) and no repeat expansion greater than 23 was detected whereas a repeat length higher than 23 was identified at the heterozygous state in 104 FALS (46%) and 57 SALS (8%) (supplementary figure 1). The frequency of the 16-23 repeat alleles was similar in ALS patients (2.9%) and control subjects (2.8%). We detected one FALS and four SALS patients with 24 repeats and two other SALS patients with 25 repeats. These intermediate repeat length carriers were not included in clinical comparison analysis. All the other positive ALS patients had GGGGCC repeat numbers greater than 50 corresponding to the detection limit of the method we used. The frequency of SALS with expansion length of more than 50 repeats was of 7%.
The segregation of the expanded repeat with the disease could be confirmed in 16 families (supplementary figure 2). The pedigrees showed that some obligate carriers were asymptomatic. Although in several families their age at death could not be determined, one of them died at 88.
The contribution of C9ORF72 gene in FALS varied according to age at disease onset: it was detected in only 17% of FALS reaching disease onset before 40 years of age (where FUS mutations are the majority) and in more than 50% of FALS with disease onset starting after 40 (figure 1A). The two groups corresponding to onset age between 41 and 60 or >61 held similar rates of 51% and 52% respectively.
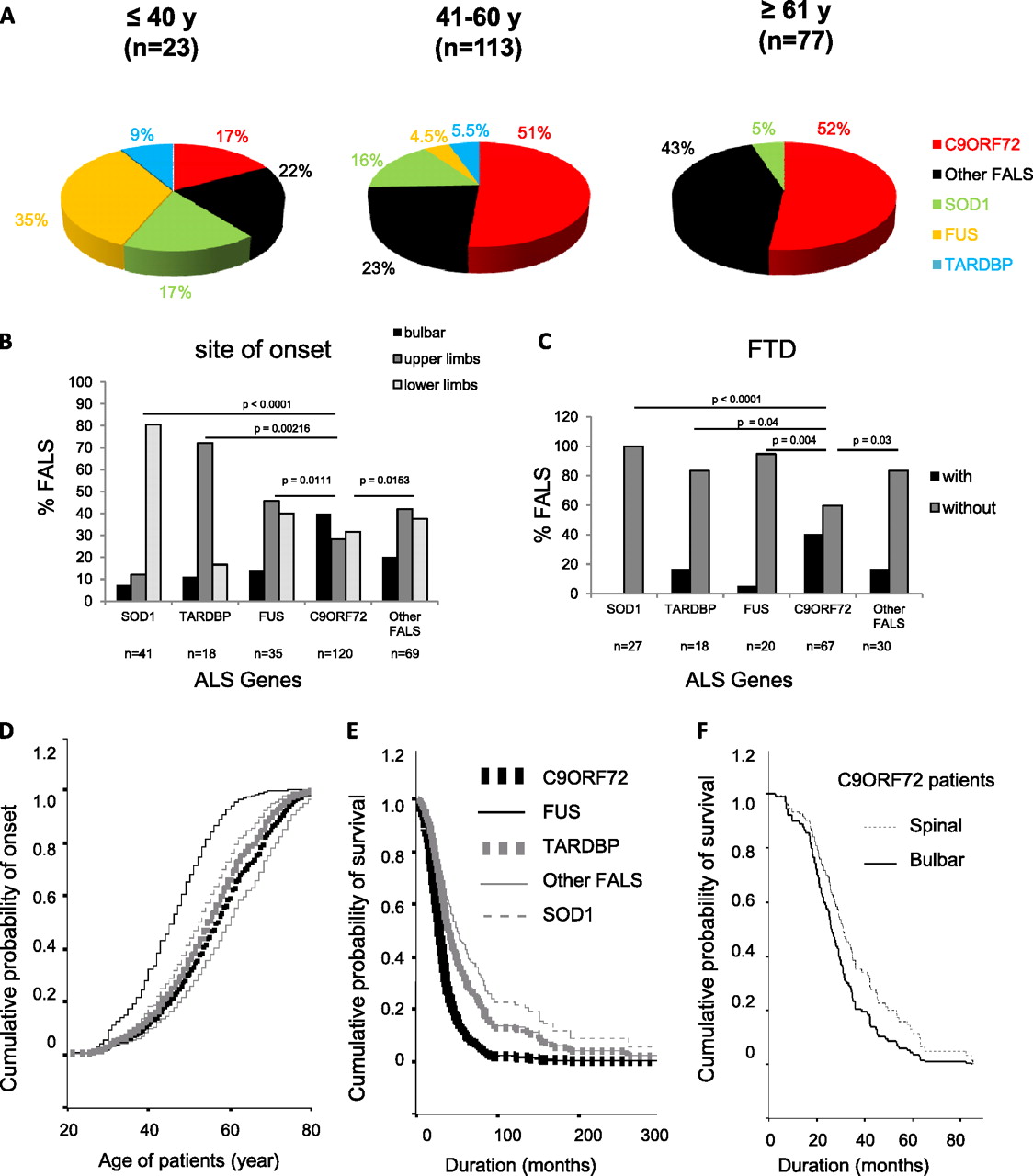
Clinical comparison of C9ORF72, SOD1, TARDPB, FUS and other FALS patients (A) Proportion of FALS patients with C9ORF72 repeat expansions, or with a mutation in SOD1, FUS and TARDBP, or with a still unidentified genetic cause (Other FALS) are presented according to age of onset: before 40 years, 41–60 years, after 61 years. (B, C) Histograms showing the distribution of FALS patients with SOD1, TARDBP, FUS mutations, C9ORF72 expanded repeats or with unidentified genetic defect (other FALS) according to bulbar (black), upper limb (dark-grey) or lower limb (light grey) onset (B) and presence (black) or absence (grey) of FTD (C). A Fischer's exact test showed a statistically significant difference between the C9ORF72 group and all the other groups of patients. Cox regression curves of cumulative probability of disease onset according to the age of FALS patients (D), and cumulative survival probability from time at disease onset (E) for C9ORF72 (black bold dotted line), FUS (black thin line), TARDBP (grey bold dotted line), SOD1 (grey thin dotted line), and other FALS (grey thin line) groups of patients. The survival curves of C9ORF72 and FUS and those of TARDBP and other FALS are superimposed. C9ORF72 and FUS patients had the shortest disease duration. Cox regression curve of cumulative survival probability from time at disease onset for C9ORF72 FALS patient with bulbar (black bold line) or spinal (black thin dotted line) onset (F). Note that disease duration was more rapid for C9ORF72 patients with bulbar onset.
Age at onset for the C9ORF72, SOD1, TARDBP, FUS and other FALS groups was statistically different (Cox regression test: p<0.001, figure 1D). C9ORF72 patients presented onset later than SOD1 (log rank: p=0.0139) and FUS patients (log rank: p<0.0001) whereas no statistically significant difference was found with the TARDBP or other FALS groups. In SALS, no difference was observed between age at onset of C9ORF72-positive and -negative patients (figure 2B).

{kind=link}
{kind=link}
Clinical comparison of SALS with or without C9ORF72 expanded repeats. (A) Histograms showing the distribution of SALS patients with (C9ORF72) or without (other SALS) C9ORF72 expanded repeats according to bulbar (black) or spinal (grey) onset. A Fischer's exact test showed a statistically significant difference between the two groups of patients. Cox regression curves of cumulative probability of disease onset according to the age (B) and cumulative survival probability from time at disease onset (C) for SALS patients who were positive (black bold dotted line) or negative (grey thin line) for C9ORF72 expanded repeats. Both curves showing probability of disease onset are superimposed. C9ORF72 patients had shorter disease duration than the other SALS.
The proportion of FALS with bulbar, lower limb and upper limb onset was different between C9ORF72 patients and the other groups (figure 1B, table 1). The site of onset was heterogeneous for C9ORF72 FALS patients, with more frequent bulbar onset (40%) than in SOD1 (7%), TARDBP (11%), FUS (14%) or other FALS (20%) patients (figure 1B). In SALS, C9ORF72 patients had also more frequent bulbar onset (40%) than the remaining ones (25%) (figure 2A).
Information about FTD was available only for a minority of our patients (figure 1C, table 1) but showed that it was more frequent in C9ORF72 FALS patients (40%) than in SOD1 (0%), TARDBP (17%), FUS (5%) and other FALS (17%) patients. A conclusion about FTD in SALS could not be drown since the information could be recovered for only 16% of the patients. The proportion of C9ORF72 FALS patients with bulbar onset was similar (41%) whether they presented or not FTD.
Disease duration differed in the five groups of FALS patients (Cox regression test: p<0.001, figure 1E). It was shorter for C9ORF72 patients than for SOD1 (log rank, p<0.0001), TARDBP (log rank, p=0.0242) and other FALS (log rank, p<0.0001) patients and similar to the FUS group (figure 1E). In SALS, disease duration was shorter for C9ORF72 patients than for the other SALS (log rank, p=0.0006; figure 2C). In C9ORF72 FALS patients, disease duration was shorter when presenting bulbar onset compared to spinal onset (log rank, p=0.04; figure 1F) but was not statistically different between patients affected or not by FDT. No gender effect was observed between C9ORF72 FALS patients and the four other groups of FALS, and between C9ORF72 SALS patients compared to the remaining SALS (tables 1 and 2).
Expanded C9ORF72 repeats were also found in our patients carrying a mutation in ANG (c.122A>T, pLys41Ile), DAO (c.113G>A, p.Arg38His), OPTN (c.382_383insAG, p.Asp128GlufsX22) or UBQLN2 (c.1500_1508delCATAGGCCC, p.Gly502_Ile504del) for whom the pathogeneicity of was questioned in previous studies.10–13 The segregation of C9ORF72 expansion could be confirmed for the family with the UBQLN2 deletion that we previously showed not to segregate with ALS phenotype (supplementary figure 2P).13 Two FALS with expanded C9ORF72 repeat (>50 repeats) also carried either a SOD1 (c.328G>T, p.Asp110Tyr, one out of the 26 index cases carrying a SOD1 mutation) or a FUS (c.1561C>T, p.Arg521Cys, one out of 13 index cases carrying a FUS mutation) mutation. Since no other affected relative was collected in these two families, the segregation of the mutations could not be further studied.
The SOD1 (p.Asp110Tyr)-C9ORF72 repeat expansion patient had upper limb onset at 59 years of age with a 42-month-disease duration. The FUS (p.Arg521Cys)-C9ORF72 repeat expansion patient had a bulbar onset at 40 and disease duration of 14 months. These patients were not included in clinical comparison analyses.
Discussion
Our results confirm that the expanded repeat in C9ORF72 is the most common genetic defect in French ALS occurring in 46% of FALS and 8% of SALS. These frequencies of C9ORF72 expanded repeats were in the range of those previously reported for FALS (23%–47%) and SALS (4%–21%) from Belgium, Finland and the USA.6–8 C9ORF72 repeat expansions were more frequent (50% of FALS) in ALS patients with disease onset starting after 40 years of age whereas FUS mutations remained the most common genetic defect (35%) in ALS patients with early onset (≤40 years). Repeat length in our control group ranged from 2 to 23, close to the findings of previous reports.6–8
Some obligate carriers were asymptomatic, suggesting either that the penetrance of the phenotype associated with the C9ORF72 expanded repeats was incomplete, or that the obligate carriers did not survive long enough to start the disease. Concerning SALS, the detection of C9ORF72 repeat expansions in these patients could be related to possible incomplete penetrance in their family, lack of complete family history or absence of clinical data regarding the dementia status of the relatives.
Comparing the phenotypes of FALS with expanded C9ORF72 repeats (>50 repeats) to the ones of patients carrying a mutation in one of the other ALS-related genes (SOD1, TARDBP, FUS) and patients with unidentified genetic defects (other FALS) we found that C9ORF72 patients had a rather late age of onset (compared to patients with other identified genetic defects). They also had more frequent bulbar onset, more frequent associated FTD and shorter disease duration than the other groups, in agreement with two other recently reported studies on smallest cohorts of patients.15 16 The proportion of C9ORF72 FALS patients with bulbar onset was similar whether they presented or not FTD implying that the site of onset and the presence of cognitive impairment did not seem to be correlated. The shorter disease duration of the C9ORF72 carrier group could be related to the more frequent bulbar presentation of the disease observed in these patients. In SALS we confirmed that C9ORF72 expanded repeat carriers had more frequent bulbar onset and shorter disease duration.
We have also found C9ORF72 repeat expansions in patients with variants in other ALS-related genes.
Thus ANG, DAO, OPTN and UBQLN2 genes appear now as negative in our cohort of 225 French FALS and should be considered as very rare causes of ALS. For patients in whom a C9ORF72 mutation was identified in addition to a potential pathogenic SOD1 mutation (SOD1 variant is an unreported substitution affecting an aminoacid that is not conserved among species) and a clearly pathogenic FUS mutation (p.Arg521Cys is one of the most frequent FUS mutation identified in ALS patients17 18), we could not perform Southern blot experiments due to limited amount of DNA for these deceased patients. As the repeat length could not be determined exactly in these patients, it is not possible to conclude if the expansion is as long as the previously reported expansions of 700 to 1600 repeat units (that were observed using Southern blot analysis6) or if in these cases, the expansion was shorter. In any case, our results suggest that the pathogenicity of any novel SOD1 missense variant should be evaluated with caution.
Since this study points out phenotype-genotype correlations between FALS groups with different ALS-related mutations, further analyses are required to define more precisely the number of repeats and to determine whether it is correlated with the severity of the phenotype (age of disease onset or disease duration) and/or the association of ALS with cognitive impairments including FTD.
Although our data have to be confirmed in other cohorts of patients, they confirm the major role of expanded repeats in C9ORF72 as causing ALS and provide evidence that these patients have a characteristic phenotype as compared to patients carrying other ALS related-gene mutations. In view of the frequency of this repeat expansion, the molecular diagnosis of ALS should be centered on the identification of this mutation.
Acknowledgments
We are grateful to the patients and their families. We thank the Généthon cell and DNA bank (Evry, France) and the CRicm DNA and cell bank (Paris, France) for patients' DNA. This work was financed by the Association pour la Recherche sur la Sclérose latérale amyotrophique et autres maladies du motoneurone (ARSla, France) and by Association française contre les myopathies (AFM, France).
References
Supplementary materials
web only data
Files in this Data Supplement:
Footnotes
Funding Association pour la Recherche sur la Sclérose latérale amyotrophique et autres maladies du motoneurone (ARSla, France) and Association française contre les myopathies (AFM, France).
Competing interests None.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the “Comité d'Ethique de la Pitié-Salpêtrière” and the Medical Research Ethics Committee of “Assistance Publique-Hôpitaux de Paris”.
Provenance and peer review Not commissioned; externally peer reviewed.