Article Text

Abstract
Electrical signals are critical for the function of neurones, muscle cells, and cardiac myocytes. Proteins that regulate electrical signalling in these cells, including voltage gated ion channels, are logical sites where abnormality might lead to disease. Genetic and biophysical approaches are being used to show that several disorders result from mutations in voltage gated ion channels. Understanding gained from early studies on the pathogenesis of a group of muscle diseases that are similar in their episodic nature (periodic paralysis) showed that these disorders result from mutations in a gene encoding a voltage gated Na+ channel. Their characterisation as channelopathies has served as a paradigm for other episodic disorders. For example, migraine headache and some forms of epilepsy have been shown to result from mutations in voltage gated Ca2+channel genes, while long QT syndrome is known to result from mutations in either K+ or Na+ channel genes. This article reviews progress made in the complementary fields of molecular genetics and cellular electrophysiology which has led to a better understanding of voltage gated ion channelopathies in humans and mice.
- ion channel genetics
- ion channel physiopathology
- channelopathies
- hereditary diseases
Statistics from Altmetric.com
Many interesting advances in molecular medicine over the last few years have come from research in molecular genetics. Virtually every month novel genes linked to different clinical disorders are cloned. Sometimes these findings relate to common diseases, while other times they concern diseases that are fairly rare. In any case, the information often provides important insight into mechanisms underlying a particular disease, or new means of understanding the function of a particular protein. A good example of this is the field of ion channel research. In parallel with the progress in the understanding of the structure and function of these proteins, the list of genetic diseases linked to them has grown rapidly. Today there are a considerable number of inherited ion channel diseases named collectively “channelopathies”, caused by mutations in K+, Na+, Ca2+, and Cl- channels that are known to exist in human and animal models.
Ion channels constitute a class of macromolecular protein tunnels that span the lipid bilayer of the cell membrane, which allow ions to flow in or out of the cell in a very efficient fashion (up to 106 per second). This flow of ions creates electrical currents (in the order of 10-12 to 10-10amperes per channel) large enough to produce rapid changes in the transmembrane voltage, which is the electrical potential difference between the cell interior and exterior. Inasmuch as Na+ and Ca2+ ions are at higher concentrations extracellularly than intracellularly, openings of Na+ and Ca2+channels cause these cations to enter the cell and depolarise the membrane. In contrast, when K+ leaves or Cl-enters the cell through open channels, the cell interior becomes more negative, or hyperpolarised. Ion channels in general can be either open or closed. The process of transition from the open to the closed state (and vice versa) is known as gating. Some channels open and close randomly at all membrane potentials. Their gating is said to be voltage independent. Other ion channels are normally closed but their open probability can be greatly enhanced by a change in membrane potential (voltage gated channels), by the binding of extracellular or intracellular ligands (ligand gated channels), or by physical stimuli (mechano and heat sensitive channels). When an ion channel opens, permeant ions are able to move through it, and the direction in which they move, as mentioned above, is governed by the electrochemical gradient that represents the sum of the chemical gradient across the plasma membrane and the electrical field experienced by the ion. Nevertheless, the movement of an ion through an open channel is not only a function of its electrochemical gradient. It is also dependent on the relative permeability of the channel to the ion, which is determined by several factors including the relative sizes of the ion and the pore of the channel. However, ion channels do not act only as molecular sieves that allow the free diffusion of ions below a certain size. Rather, they can discriminate in the kind of ions to which they are permeable. For example, Na+ channels are highly permeable to Na+ but not to K+ ions, whereas K+ channels are ∼100 times more permeable to K+ than to Na+. Because Na+ has a smaller ionic radius than K+, the high selectivity of the channels cannot be simply explained by physical occlusion. This ionic discrimination takes place where the pore of the channel is narrowest, at a region known as the selectivity filter, and it is the amino acids located at the selectivity filter that determine which ions can permeate. Cation selective channels, for example, often have negatively charged residues at, or near, their selectivity filters, which attract positive ions and repel negative ions.1 2
This review focuses on the voltage gated ion channels for cations (Na+, K+, and Ca2+), and briefly discusses several recent studies linking a growing number of genetic disorders to genes of the ion channel superfamily, with special emphasis on those characterised by neurological, neuromuscular, or cardiac dysfunction in humans and mice. However, in order to be able to put the information on channelopathies in perspective, it is necessary to consider first some molecular aspects of voltage gated ion channel structure and function.
Voltage gated ion channels and cellular excitability
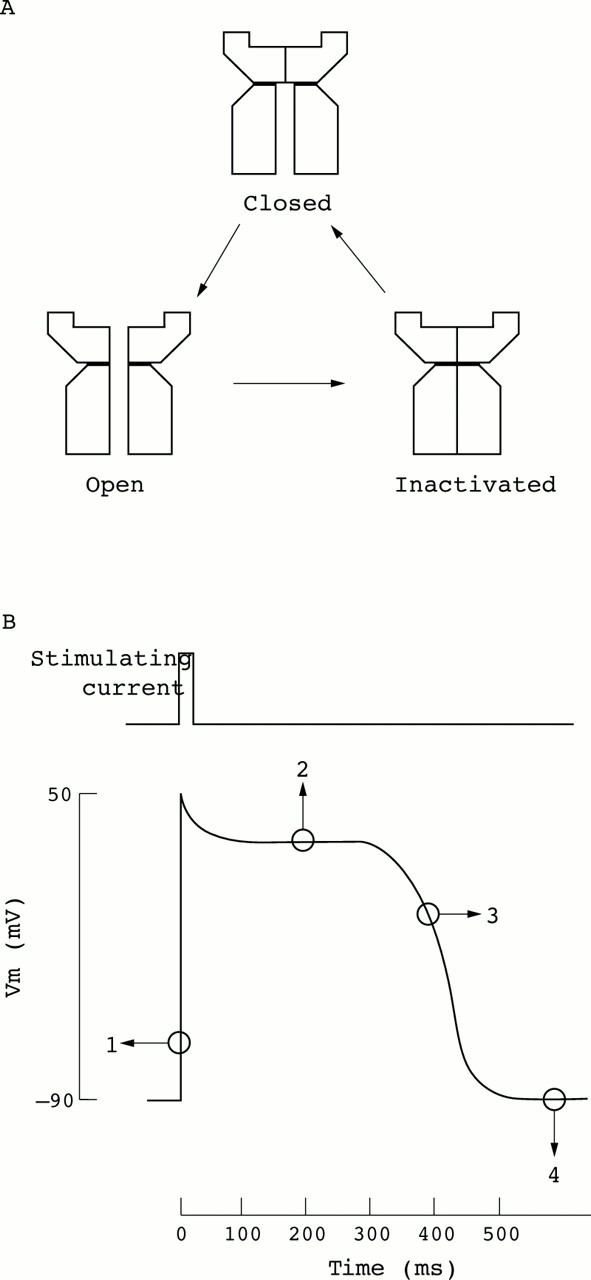
Voltage gated ion channels can assume either of three major conformational states, closed, open, or inactivated, and usually respond to membrane depolarisation with a transition from the closed to an open state followed by an intrinsic inactivation (fig1A).1 2 Na+ and K+ channel activation and inactivation are the basis of the action potential, the principal integrating signal that allows excitable cells to conduct information for the control of a wide range of physiological events including, among others, propagation of the nerve impulse and cardiac pacemaking (fig 1B). Furthermore, Ca2+ channels not only contribute to membrane polarisation per se, but also play an additional critical role in controlling the influx of this important intracellular regulator. Once within the cell, Ca2+ interacts with modifying enzymes, cofactors, and other second messengers to influence cellular events ranging from muscle contraction, exocytosis, and enzyme activation to gene expression.2 3 Voltage gated ion channels may also participate in the regulation of many specific functions in non-excitable cells. For example, in mammalian sperm capacitation, a poorly understood maturational process, a modification of K+ channel activity, occurs that render sperm responsive to stimuli leading to the acrosome reaction before fertilisation.4

Ion channels and electrical excitability. (A) Plasma membrane voltage gated channels can adopt at least three conformational states. At rest, the closed (but not inactivated) state has the lowest free energy and is therefore most stable; however if the membrane is depolarised, the energy of the open state is lower and the channel opens allowing ions to flow. The cell is saved from a permanent spasm because the free energy of the inactive state is lower still, and after a randomly variable period spent in the open state the channel becomes inactivated (stops conducting). In this state the channel cannot open again until the membrane potential returns to its initial negative value. (B) Voltage gated ion channels are responsible for generating transient self-propagating electrical signals called action potentials. The time course of a prolonged cardiac action potential is illustrated. In this particular case, an excitatory stimulus that causes a momentary partial depolarisation beyond a threshold voltage promptly causes voltage gated Na+ channels to open (1) causing further depolarisation of the cell membrane. Na+ channels are then rapidly inactivated allowing a transient K+ current to return the action potential to a plateau voltage (2). This plateau is maintained by the balance between outward and inward moving currents through K+ and Ca2+ channels, respectively. Progressive inactivation of Ca2+ channels and increasing activation of K+ channels repolarise the cell (3) to the resting membrane potential (4), which is maintained by inwardly rectifying K+ channels.
Molecular structure of voltage gated ion channels
As mentioned above, ion channels have distinct ion selectivity and gating properties, determined at the molecular level by the type of the pore forming (α) subunit that the channel contains (fig 2A). The structure of the α subunits is modelled as six hydrophobic segments (S1-S6) embedded in the plasma membrane with the N- and C-terminal domains of the protein positioned intracellularly. One transmembrane segment (S4) contains a unique array of positive charges that function as the voltage sensor of the channel. The region separating segments S5 and S6 may contain two segments that together form the pore of the channel.1 2

Structure and function of voltage gated K+ channels. (A) Voltage gated K+ channels of the Shaker related superfamily are assembled from membrane integrated α subunits and auxiliary β subunits. The β subunits may increase K+ channel surface expression and confer native kinetic behaviour to α subunit K+ channels expressed in heterologous systems. (B) A functional voltage gated K+channel is formed by four α subunits clustered around the central pore. A tetrameric channel complex can be formed by physical association of identical (homomers) or different (heteromers) subunits. (C) The most recently discovered family of ion channels is that containing the inwardly rectifying K+ selective channels (IRKs). The general structure of an IRK channel consists of two membrane spanning domains (M1 and M2) that flank a highly conserved pore (P) region containing the conserved H5 segment (upper panel). The H5 and M2 segments, in conjunction with the carboxyl-terminus hydrophilic domain, are critical for K+ permeation. As in Shaker-like channels, four channel subunits presumably assemble to form a functional IRK channel. This group of K+ channels conducts current (IK) much more effectively into the cell than out of it and determine the transmembrane voltage (Vm) of most cells at rest (see also fig 1B), because they are open in the steady state (lower panel). (D) Using positional cloning methods to establish a gene, it was possible to identify the gene (KVLQT1) responsible for long QT syndrome. KVLQT1 is strongly expressed in the heart and encodes a protein with structural features of a voltage gated K+channel. (E) The voltage gated K+ channel KCNQ1 associates with the KCNE1 subunit (a small one transmembrane segment protein, right panel) to form the slow cardiac outward delayed rectifier K+ current (IKs). KCNQ1 can form functional homotetrameric channels, but with drastically different biophysical properties compared to heteromeric KCNQ1/KCNE1 channels. A great deal of understanding of cardiac arrhythmogenesis is emerging as the function of these channels has been elucidated. In fact, mutations in both subunits can cause long QT syndrome (listed in table 1). (F) The HERG K+ channel is atypical in that it seems to have the structure of the voltage gated K+ channel family (six putative transmembrane segments), yet it exhibits rectification like that of the inward rectifying K+ channels, a family with different molecular structure (two transmembrane segments, illustrated in C). Functional studies on HERG channels have suggested that the inward rectification arises from a rapid and voltage dependent inactivation process that reduces conductance at positive voltages.
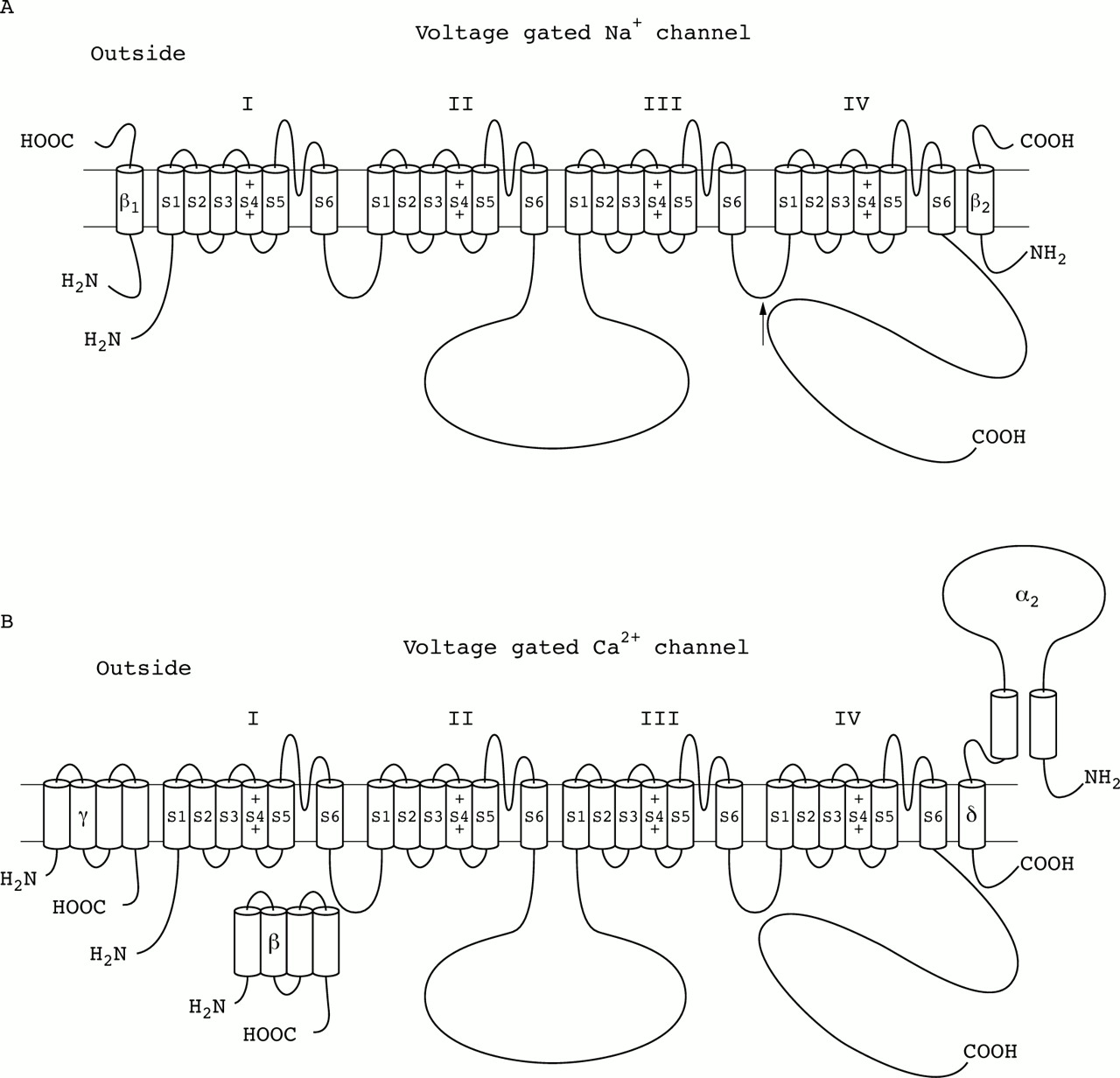
Most of the functional K+ channels are formed when four α subunits aggregate to produce a hetero- or homotetrameric structure (fig 2B). However, evidence exists that some K+ channels more distantly related to this superfamily of voltage gated channels (for example, the inwardly rectifying and the G protein regulated inwardly rectifying K+ channels, IRKs and GIRKs, respectively) possess only two membrane spanning segments that resemble the S5 and S6 regions of the α subunit (fig 2C). Functional Na+ and Ca2+ channel α subunits contain four sets of S1-S6 membrane spanning regions coded as parts of a single gene product linked by hydrophilic loops. Even though the α subunits are capable of conducting ions by themselves, voltage gated ion channels often include one or more ancillary proteins called “auxiliary subunits” that play modulatory, structural, or stabilising roles.5 These auxiliary subunits can be subdivided into two main classes. One class consists of the entirely cytoplasmic intracellular subunits, which have no transmembrane domains such as the β subunits of the voltage gated K+ (fig 2A) and Ca2+ channels (fig 3B). The other class of auxiliary subunits contains at least one transmembrane domain. These proteins include the α2δ and γ subunits of the Ca2+ channel (fig 3B) and the β subunits of the Na+ channel (fig 3A).6 Most traditional voltage gated K+ channels have not been shown to have transmembrane containing subunits, although a family of membrane proteins including IsK (minK) may fit into this category (fig 2E).

{kind=link}
{kind=link}
{kind=link}
Subunit structure of the voltage gated Na+and Ca2+ channels. (A) Na+ channels are responsible for action potential generation in most excitable cells and consist of a complex of three glycoprotein subunits: a pore forming α subunit of ∼260 kDa associated non-covalently with a β1subunit of ∼36 kDa and disulphide linked to a β2subunit of ∼33 kDa. The α subunit forms a functional channel by itself, and is composed of four homologous domains (I-IV), which contain six probable helical transmembrane segments (S1-S6) and an additional membrane associated pore loop. β1 and β2 subunits are single membrane spanning glycoproteins that modulate channel gating. Structure-function analyses of the α subunit have shown that the S4 transmembrane segments in each domain serve as voltage sensors for channel activation, the S5 and S6 segments and the pore loop between them form the transmembrane pore, and the highly conserved intracellular loop between domains III and IV is proposed to form the inactivation gate (arrow). The β auxiliary subunits do not form the pore but play important roles in modulating channel function, as well as plasma membrane expression levels. (B) Ca2+ channels provide a crucial link between a cell's membrane potential and an enormous number of intracellular processes that either directly use increases of [Ca2+]Ias a functional trigger (for example, exocytosis, muscle contraction) or are modulated by Ca2+ dependent signalling cascades (for example, gene expression, cell division). These channels are complexes of a pore forming α1 subunit, a transmembrane δ subunit disulphide linked to an extracellular α2 subunit, an intracellular β subunit, and, at least in some tissues, a γ subunit. Expression of cloned α1 genes has shown many functional roles for this subunit, including sensing of the transmembrane voltage, channel activity, and binding of pharmacological agents. As in voltage gated Na+ channels, the α1 subunit (∼190-250 kDa) of Ca2+ channels is composed of four homologous membrane spanning internal domains, each with six transmembrane helices and a pore forming re-entrant P loop. Carboxyl- and amino-termini and linkers between domains I-II, II-III, and III-IV are cytosolic. The α2δ complex (∼170 kDa) derives from a common precursor protein that is proteolytically processed to yield separate α2 and δ proteins that remain linked by disulphide bonds. This auxiliary subunit determines membrane localisation and modifies distinct biophysical and pharmacological properties of the ion conducting α1subunit. The β ancillary subunits are cytoplasmic proteins (of ∼50-80 kDa), which facilitate the incorporation of channels into the plasma membrane and modulate Ca2+ currents. The γ subunit is a ∼26 000 dalton glycosylated hydrophobic protein that is predicted to contain four transmembrane domains. Functional studies suggest that γ is important in channel assembly and modulates channel properties in a subtype specific manner.
Human channelopathies and mouse models
K+ CHANNELOPATHIES
Episodic ataxia type 1 (EA1) is the only human ataxia known to be caused by dysfunction of a K+ channel (table 1). Some other types of this neurological disorder are associated with mutations in Ca2+ channels (see below). EA1 is a rare, disabling condition of autosomal dominant inheritance. Although family members have similar clinical features, the syndrome varies considerably from family to family. The disorder begins in early childhood with attacks of ataxia of one to two minutes in duration, with associated interictal uncoordinated movements of the head, arms, and legs. Attacks are provoked by abrupt postural change, emotional stimulus, and caloric vestibular stimulation. EA1 is the result of different heterozygous missense point mutations in a neuronal voltage gated (delayed rectifier) K+ channel gene (KCNA1/Kv1.1) on chromosome 12p13 (table 1).7 Expression inXenopus oocytes showed that two of these EA1 subunits are indeed able to form functional channels, though their gating properties are altered. A Val408 to Ala mutation produces channels with voltage dependence similar to that of wild type channels, but with faster kinetics and increased C type inactivation (a slow inhibition in channel activity induced by voltage), while the voltage dependence of Phe184 to Cys mutant channels is shifted 20 mV in the depolarising direction. Several other EA1 subunits have failed to produce functional homomeric channels, but reduced the K+current when coassembled with wild type subunits.8 Because studies in Xenopus oocytes indicate that the mutations causing EA1 result in reductions inKCNA1 expression and current amplitude, knockout mice lacking the murine homologue ofKCNA1 became a useful animal model for EA1. Mice heterozygous for the null allele appear normal while homozygous mutants have very frequent generalised epileptic seizures.9
K+ channel genes associated with neurological and cardiac disorders in human and mouse
Extensive further work has provided useful clues for understanding the basis of the EA1 phenotype. By using specific antibodies, it was found that kcna1 is localised to a key site regulating cerebellar output, the axons and nerve terminals of the basket cells that make inhibitory synaptic contacts on Purkinje neurones.10 Patch-clamp recordings of these cells showed an extraordinarily high K+ channel density. Blocking these channels with α-dendrotoxin caused a dramatic increase in the spontaneous inhibitory postsynaptic potentials recorded in Purkinje cells, suggesting that kcna1 regulates the excitability of the basket cell presynaptic terminals.11
To date, all inherited arrhythmogenic disorders in which the causative genes have been identified turned out to be channelopathies, since the genes encode channel subunits that regulate important ion currents that tune the cardiac action potential. Congenital long QT syndrome was the first genetically determined arrhythmia shown to be caused by defects in myocardial ion channels. This clinical condition involves an alteration characterised by prolongation of the QT interval of the electrocardiogram, which results from an increased duration of the cardiac action potential. Most patients suffering from long QT syndrome are asymptomatic and only one third of the cases are identified by electrocardiographic screening during a clinical evaluation for either unexplained syncope or cardiac or respiratory arrest. Often the arrhythmia is a polymorphic ventricular tachycardia known as torsade des pointes, triggered by adrenergic arousal or by exposure to a variety of medications.12 13 Recent studies have implicated several genetic defects in KCNQ1(table 1), a 581 amino acid protein with sequence homology to the α subunit of voltage gated K+ channels (fig 2D) in the aetiology of the disease.14-16
The association of this KCNQ1 channel protein with another subunit named IsK (also known as minK) initiates the IKs current (fig 2E), which is the principal delayed rectifying K+ current responsible for repolarising the cardiac myocytes during the action potential.17 Mutations in the K+ channel gene (KCNQ1) and the consequent alteration of the IKs current results in sustained depolarisation and abnormally long cardiac action potentials. Similarly, physiological studies showed that two mutations in theKCNE1 gene encoding the human minK subunit significantly reduced IKs current by altering some of the fundamental biophysical properties of the channel including its voltage dependence of activation and kinetics of inactivation.18The main functional consequence of these mutations would be delayed cardiac repolarisation and an increased risk of arrhythmia.
Mutations in a third K+ channel known asHERG (human ether-a-go-go-related gene, fig2F) have been identified in subjects suffering from a second form of the long QT syndrome (type 2). HERG is responsible for another major K+ current IKrthat participates in the repolarisation of the cardiac action potential. Although HERG has the secondary structure of a typical voltage gated K+ channel (fig 2A), it behaves like an inward rectifier because it conducts current much more effectively into the cell than out of it. The role of this channel in cardiac physiology appears to be to suppress depolarisations that lead to premature firing. Patients with long QT syndrome type 2 may therefore be prone to sudden cardiac death, because they lack protection from arrhythmogenic afterbeats.19 20
The discovery of the genetic bases of the LQT syndrome became a new methodological paradigm. With the use of genetic linkage strategies, not only have the causative genes been found, but functional components with a previously unknown but fundamental role for a normal repolarisation process have been discovered.
Two rare forms of epilepsy known as benign familial neonatal convulsions (BFNC) type 1 and 2 have been linked to mutations in two novel and related genes (KCNQ2 andKCNQ3, respectively) coding for the voltage gated K+ channel α subunit (table 1). These forms of epilepsy are characterised by frequent, brief seizures after the second day of life which often disappear spontaneously within weeks. Newborns show normal behaviour between seizures and normal subsequent neurodevelopment. The seizure phenotype usually starts with a tonic posture and shallow breathing, followed by ocular signs (staring, blinking, or gaze deviation), clonic movements, and automatisms. Electroencephalograms recorded during seizures show a characteristic sequence of alterations that correlate with clinical signs. In the case of BFNC1, the genetic defects include two missense, two frameshift, and one splice site mutations, while in patients with BFNC2 the alteration comprises a single missense mutation in the pore region of the channel (table 1). Since voltage gated K+ channels are responsible for repolarising the cell membrane during the action potential, and they are also thought to participate in membrane repolarisation after activation of excitatory neurotransmitter ion channels, it has been speculated that in the presence of mutant K+ channels with reduced function, both voltage and ligand gated ion channels would remain open longer.21 22 Recently, Wanget al 23 reported that heterotetramers consisting of KCNQ2 andKCNQ3 form the so called M channels that regulate the subthreshold electrical excitability of neurones. Although it is likely that BFNC is caused by neural hyperexcitability owing to dysfunction of M channels, there has been no direct evidence that an alteration in a KCNQ gene can actually cause epilepsy.23 However, mice heterozygous for the null allele of the KCNQ2 gene showed normal behaviour and morphology compared with wild type mice, but displayed increased sensitivity to an epileptic inducer.24
Lastly, two more targeted mutations in the murine orthologues of the α subunit of K+ channels have been described. Their phenotypes vary from seizures to more subtle signs, including poorly coordinated motor skills and inner ear defects.25 26Perhaps the best studied K+ channel linked disorder in mouse is the weaver (wv) mutation, which results from a genetic defect in the KCNJ6gene encoding the G protein coupled inwardly rectifying K+channel (GIRK2).27 28 Weaver mice are characterised by male infertility, spontaneous seizures, extensive cerebellar granule cell death, and progressive loss of substantia nigra neurones. Neuronal death in weaver has also been attributed to the loss of G protein activated inwardly rectifying K+ currents in animals carrying the mutant allele.29 Because of the postnatal degeneration of dopaminergic neurones exhibited bywv/wv mutant mice, weaver also provides an interesting animal model for Parkinson disease.
NA+ CHANNELOPATHIES
The periodic paralytic disorders of skeletal muscle and the non-dystrophic myotonias are diseases resulting from mutations in voltage gated Na+ channels (fig 3A). In particular, hyperkalaemic periodic paralysis (HyperPP), paramyotonia congenita (PMC), acetazolamide responsive myotonia, and myotonia permanens/fluctuans are all clinically distinct autosomal dominant disorders associated with mutations in the α subunit of Na+ channels.
HyperPP is an autosomal dominant disorder characterised by episodes of muscle weakness resulting from depolarisation of the muscle cell membrane associated with raised serum K+. Early molecular studies found that the adult muscle Na+ channel α subunit gene (SCN4A) contained the HyperPP mutation.30 Analysis of DNA from unrelated patients with HyperPP showed a C to T transition, predicting a substitution of a highly conserved threonine residue with a methionine in a membrane spanning segment of the Na+ channel protein.31Further genetic analysis of the α subunit coding region from affected HyperPP patients identified several mutations that cause changes in both the highly conserved transmembrane regions and the variable cytoplasmic domains of the Na+ channel α subunit (table2). Electrophysiological data indicate that Na+ channels from muscle cells of HyperPP affected subjects show abnormal activation32 and inactivation.33 34
Na+ channel genes associated with neurological and cardiac disorders in human and mouse
Similarly, several mutations that occur in the adult skeletal muscle voltage gated Na+ channel SCN4A gene (table 2) have been identified as causing paramyotonia congenita (PMC), a temperature sensitive disorder characterised by exercise and cold induced myotonia and weakness. Recent studies have emphasised the role of channel mutations that cause a positive charge decrease in segment S4 of domain IV of the ion conducting α subunit. In particular, a single nucleotide substitution causing an amino acid change at position 1448 in segment S4 of domain IV has been implicated in the phenotype of PMC. Interestingly, expression of the altered proteins in HEK293 cells showed several defects in channel function including alterations in the kinetics and voltage dependence of inactivation, slower deactivation, and faster recovery from inactivation.35-38
Ricker et al 39 discovered a novel mutation in exon 22 of the Na+ channelSCNA4 gene, which encodes the region containing the cytoplasmic loop between domains III and IV. A G to C transition was found at position 3917, predicting a Gly1306 to Ala substitution. This mutation was found in affected members of families with the myotonia fluctuans phenotype.39 This consists of fluctuating myotonia of varying severity, a warm up phenomenon, worsening of myotonia after K+ loading, increased myotonia with delayed onset following exercise, and no significant increase of myotonia following exposure to cold. Similarly, one missense mutation (I1160V) that occurs at a very highly conserved position in the Na+ channel α subunit has been reported as the putative disease causing mutation in acetazolamide responsive myotonia congenita,40 41 an autosomal dominant, painful myotonia that is K+ sensitive and responsive to acetazolamide.
Myotonic muscles recorded in vivo exhibit abnormal spontaneous oscillations in membrane potential, called myotonic discharges. In normal muscle, depolarisation of the postsynaptic membrane causes the opening of Na+ channels within the first few msec after depolarisation. As these Na+ channel openings subside, Cl- enters the cell through more slowly opening Cl- channels, promptly returning the membrane potential to its resting level. Na+ channels harbouring mutations causing myotonia exhibit an abnormal tendency to open later or more persistently after membrane depolarisation. Residual Na+entry through these abnormal channels repeatedly reinitiates the cycle of membrane depolarisation.42
As mentioned above, heritable long QT syndrome is a disease in which delayed ventricular repolarisation leads to cardiac arrhythmias and the possibility of sudden death. Wang et al 23 mapped three LQT loci:LQT1 on chromosome 11p15.5,LQT2 on 7q35-36, andLQT3 on 3p21-24. They reported genetic linkage betweenLQT3 and polymorphisms withinSCN5A, the cardiac Na+ channel gene. Single strand conformation polymorphism and DNA sequence analyses showed intragenic deletions of SCN5A in affected LQT patients, indicating that mutations inSCN5A cause chromosome 3 linked LQT.43 In the disease, one mutation of the cardiac Na+ channel α subunit gene results in a deletion of residues 1505 to 1507, and two mutations result in substitutions (N1325S and R1644H). These altered sequences reside in a region that is important for channel inactivation, suggesting a likely cellular mechanism for the cardiac disorder. LQT3mutant channel expression in Xenopus oocytes produced inactivation resistant whole cell currents.44-46
A transgene induced intragenic deletion of the brain Na+channel Scn8a has been identified in mice with motor end plate disease (med), a genetic defect that causes progressive neuromuscular failure. Mice homozygous for this mutation display altered cerebellar function and progressive neuromuscular weakness, which begins ∼10 days after birth and results in death within 3-4 weeks.47 Analysis of homozygousmed mice showed that the development and function of mature motor neurones depends on the postnatal induction of Scn8a expression.48 Similarly, Kohrmanet al 49 showed that a mutation that shifts the voltage dependence of activation of the channel is associated with inherited cerebellar ataxia in the “jolting” mouse.49 This mutant mouse arose as a spontaneous mutation and genetic mapping and complementation tests showed that jolting is a mild allele (med jo) of the motor endplate disease locus (med) on mouse chromosome 15 that exhibits cerebellar ataxia only.50 The jolting mutation results in substitution of Thr for a conserved Ala residue in the cytoplasmic S4-S5 linker of domain III in the α subunit of the channel (fig 3A). Homozygous jolting mice have ataxia of cerebellar origin and a rhythmical tremor of head and neck. Electrophysiological studies have shown that the absence of spontaneous, regular, simple discharges from the Purkinje cells in the cerebellum of med jo mice, and the introduction of the jolting mutation into the rat brain IIA Na+ channel, expressed inXenopus oocytes, shifted the voltage dependence of activation in the depolarising direction, providing the molecular basis for the reduced spontaneous activity of Purkinje cells, reduced inhibitory output from the cerebellum, and loss of motor control observed in the jolting mouse.51
CA2+ CHANNELOPATHIES
Molecular biological analyses have identified a gene family of α1 subunits (A, B, C, D, E, F, G, H, I, and S) encoding six functionally different types (L, N, P, Q, R, and T) of Ca2+ channels (fig 3B). Ophoff et al 52 identified mutations in the human Ca2+ channel α1A subunitCACN1A4 gene in episodic ataxia type 2 (EA2) and familial hemiplegic migraine (FHM) patients. EA2 is an autosomal dominant paroxysmal cerebral disorder, characterised by acetazolamide responsive attacks of ataxia and migraine-like symptoms, interictal nystagmus, and cerebellar atrophy. FHM is a rare autosomal dominant subtype of migraine with aura, associated with ictal hemiparesis and, in some cases, progressive cerebellar atrophy. In EA2, two mutations disrupting the reading frame were originally found, one base pair deletion (C4073) resulting in a truncated Ca2+ channel α1 subunit and a G to A transition of the first nucleotide of intron 24 (table 3). Four different missense mutations were identified in FHM patients: a transition from G to A at codon 192, leading to substitution of an Arg for Glu (R192Q) within the fourth segment of the first membrane spanning domain (IS4); a missense mutation at the second repeat, replacing a Thr with Met (T666M); and two mutations located in the sixth transmembrane spanning segment of repeats II and IV (V714A and I1811L, respectively). Functional studies provided evidence that three of these four mutations affect the kinetic properties and the voltage dependence of activation of the α1A Ca2+ channel.53 In addition, a locus for spinocerebellar ataxia type 6 (SCA6), which is another autosomal dominant paroxysmal cerebral disorder, was mapped to chromosome 19p13.1 in the same interval as the EA2 and FHM locus,54 55 indicating that EA2, FHM, and SCA6 are allelic Ca2+ channel disorders. Histological studies showed neuronal degeneration confined mainly to the cerebellar Purkinje cells,56 suggesting a likely pathological mechanism for the neurological phenotype. Interestingly enough, a polymorphic CAG repeat was identified in the human Ca2+ channel α1A subunit,57 58 providing the molecular basis for this disorder.
Ca2+ channel genes associated with neurological and neuromuscular disorders in human and mouse
Incomplete X linked congenital stationary night blindness (CSNB2) is a recessive non-progressive retinal disorder characterised by night blindness, decreased visual acuity, myopia, nystagmus, and strabismus. Electrophysiological data suggested a defect in retinal neurotransmission, while molecular studies localised theCSNB2 locus to chromosome Xp11.23.59 Interestingly, a Ca2+ channel α1 subunit gene (CACNA1F), which shares high homology with the dihydropyridine (DHP) sensitive L type channel was identified in this region.60 61 Genetic analysis of this novel gene in CSNB2 patients showed several different mutations (table 3), including nonsense and frameshift mutations that would be predicted to cause premature protein truncation, suggesting that aberrations in this voltage gated Ca2+ channel cause this disorder.59-61
Hypokalaemic periodic paralysis (HypoPP) is an autosomal dominant muscle disease manifested by episodic weakness associated with low serum K+. It is thought to arise also from the abnormal function of Ca2+ channels. Fontaine et al 62 localised the HypoPP locus to chromosome 1q31-32 and showed that the gene encoding the skeletal muscle DHP sensitive Ca2+ channel α1S subunit (CACN1A3) maps to the same region. Ptacek et al 63 showed that mutations in theCACN1A3 gene causing a substitution of a highly conserved Arg in the S4 segment of domain IV in α1S with either His or Gly are associated with HypoPP. Functional expression of these mutant subunits, as well as a third mutation in the S4 segment of domain II (R528H), inXenopus oocytes showed reduced L type Ca2+ current amplitude and altered activation properties.64 Although this mutation induced only minor differences in the electrophysiological properties of α1S, it significantly reduced the whole cell Ca2+ current in its amplitude when expressed in mouse Ltk- cells.65
Initial studies in subjects suffering from malignant hyperthermia susceptibility (MHS), a potentially fatal autosomal dominant disorder of skeletal muscle triggered in susceptible people by inhalation anaesthetics, indicated that a mutation in an intracellular Ca2+ releasing channel, the ryanodine receptor type 1 (RYR1), was linked to the disease.66 However, the demonstration of genetic heterogeneity in MHS67 prompted the investigation of the roles played by Ca2+ regulatory proteins other than RYR1, which was known to be linked to MHS in fewer than half of the cases.68 In several MHS families, Ileset al 69 found linkage with no recombination to markers flanking the CACN2Alocus on chromosome 7. Since this gene encodes the α2δ subunit of the L type voltage gated Ca2+ channel that is intimately associated at the skeletal muscle triadic junctions with the RYR1, it was thought that the mutation was located in this gene. This, and the recently discovered mutations in the β4 subunit gene (see below) in affected subjects with idiopathic generalised epilepsy and hereditary episodic ataxia, are the only direct implication of an auxiliary Ca2+ channel protein (fig 3B) in human genetic disease.70 Although detailed analysis of the sequence and genomic structure of theCACNA2 gene have been performed, mutations within the coding region of this gene have not yet been identified.71 However, since the promoter region remains to be analysed, and the eventuality of an intronic mutation is still possible, CACNA2 remains a prime candidate for MHS in subjects who show no mutation in the Ca2+channel ion conducting subunits of skeletal muscle. How mutations in either RYR1 or the Ca2+ channel α2δ subunit might result in the same disease remains unknown. However, both Ca2+ channels are involved in excitation-contraction coupling and have been shown by electron microscopy to be in close approximation to each other in skeletal muscle.72 Thus, the possibility exists that mutations in the α2δ subunit could influence the behaviour of the α1S subunit as a voltage sensor for excitation-contraction coupling. More recently, evidence suggesting two further loci for MHS was published.73 One of these is located in chromosome 1q, the site of a candidate gene,CACN1A3, encoding the α1Ssubunit of the DHP sensitive skeletal muscle L type Ca2+channel. The second region resides on chromosome 5p where no known candidate has been mapped yet. Sequence analysis of the coding region of the CACN1A3 gene showed the presence of an Arg-His substitution at residue 1086, resulting from the transition of A to G3333, which segregates with the MHS phenotype.74
As mentioned above, very recently two mutations have been reported in the human β4 subunit gene (R482X which results in a protein truncated in the middle of the domain that interacts with the C-terminus of the α1 subunit, and a GT transversion resulting in the replacement of a Cys residue 104 by Phe) cosegregate with the disease in affected subjects suffering from idiopathic generalised epilepsy and episodic ataxia.70 However, the alterations of the α1A Ca2+ currents caused by the functional expression of these mutant subunits in the oocyte expression system were too small to support linkage.70
Similarly, various spontaneous mutants and natural strain variants for either generalised tonic-clonic seizures or non-convulsive absence seizures have been described in mice over the years. Some of these abnormalities have been attributed to specific alterations of Ca2+ channel activity. Hence, a missense mutation was found in the voltage gated Ca2+ channel α1A gene in the tottering (tg) mice, which display a delayed onset, recessive disorder consisting of ataxia, motor seizure, and absence seizure resembling petit mal epilepsy. Thetg mutation causes a Leu-Pro substitution at a position close to the conserved pore lining region (P region) in the extracellular segment of repeat II. Mice with an allelic tottering mutation leaner (tg la), which causes more severe symptoms, were found to have a single nucleotide substitution at an exon/intron junction, which results in skipping of the exon, or results in failure to splice out the succeeding intron. In both cases, the tg la mutation causes truncation of the normal open reading frame and expression of aberrant C-terminal sequences.75 Comparison of the properties of Ca2+ channel current in Purkinje cells of normal and tottering mutant mice showed a reduction of current density76 77 and changes in channel gating, which could be reproduced with recombinant mutant channels expressed in a mammalian cell line.78 A third additional tottering mutation, found in rolling Nagoya (tg rol) mice, manifests with poor motor coordination leading to falling and rolling, and in some cases stiffness of the hindlimbs and tail, but no motor seizures. Recently, Mori et al 79 found that thetg rol mutation leads to a charge neutralising Arg to Pro substitution in the voltage sensor forming segment S4 in repeat III of the α1A subunit. Electrophysiological analysis of α1A Ca2+channels in tg rol mutant mouse indicated decreased currents and deviation in the voltage sensing mechanisms of the channels.80
In addition to these mutations in the α1A pore forming subunit of the P/Q type Ca2+ channels, alterations in three mouse models have been associated with mutations in Ca2+channel auxiliary subunits. The mouse mutant lethargic(lh) exhibits severe neurological defects including ataxia, episodic dyskinesia, and generalised spike-wave epilepsy owing to a mutation that deletes the α1 subunit interaction domain of the β4 subunit. Specifically, thelh mutation causes a 4 bp insertion into a splice donor site within the Cacnb4 gene on chromosome 2.81 The mutation results in aberrant pre-mRNA splicing and translational frameshift and is predicted to encode a severely truncated β4 protein missing 60% of the C-terminus relative to wild type including, as mentioned above, the essential α1-β interaction domain. Notably, β subunits expressed with deletions of this domain lose their ability to modulate α1 subunit function in vitro.82More recently, it has been documented that neither full length nor truncated β4 protein are expressed as a result of thelh mutation.83
While attempting to identify a new human tumour suppressor gene in chromosome region 3p21.3, Gao et al 84 identified a novel gene that encodes a homologue of the α2δ subunit of Ca2+channels (CACNA2D2). Northern analysis showed that the CACNA2D2 gene is well expressed in brain, and in vitro studies showed an increased number of functional channels at the plasma membrane after cRNA co-injection with α1B recombinant Ca2+ channels in theXenopus oocyte expression system.84 Interestingly enough, the human orthologue of this novel Ca2+ channel α2δ auxiliary subunit has been shown to map to the ducky (du) candidate region of mouse chromosome 9.84 85 du is a spontaneous autosomal recessive mutation that is thought to be a valid model of human idiopathic generalised epilepsy. Homozygousdu mice also display ataxia, hind brain dysgenesis and demyelination, and axonal dystrophy of selected nerve fibres.86 Lastly, in the third mouse model, the stargazer (stg) mutation causes absence seizures that are more prolonged and frequent than any other petit mal mouse model.stg mice also have an ataxic gait and vestibular problems, including a distinctive head tossing motion. The stargazer locus was mapped between D15Mit30 and the parvalbumin gene, and a novel gene whose expression is disrupted in two stargazer alleles was characterised.87 88 This gene,Cacng2, was shown to encode the first of several γ subunits recently discovered in the nervous system.88-91 Transient transfection of γ2in BHK cells stably expressing recombinant α1A (P/Q type) Ca2+ channels decreased the availability of the channels, as indicated by a negative shift in the inactivation curve for the channels in the steady state.89 Inasmuch as P/Q type channels are major mediators of neurotransmitter release at the presynaptic terminals,92 it was speculated that γ2 would inhibit presynaptic Ca2+ entry instg mutant mice, causing an inappropriate modulation of an inwardly rectifying cationic current (Ih) which normally governs excitability in thalamic neurones.93 94 In this manner, the upregulation of Ih in stg would be a likely mechanistic link between the mutation and the epileptic phenotype.89 94
Conclusion
Although biophysical studies of mutant voltage gated ion channels in vitro allow detailed investigations of the basic mechanism underlying channelopathies, a full understanding of these diseases requires knowing the roles these channels play in their cellular and systemic context. It should be noted that in many cases the mutations have been identified but the mechanism by which the mutation causes the abnormal phenotype is unclear. For example, the molecular basis of the tottering mouse phenotype was identified as a point mutation within the α1A subunit of the P/Q type voltage gated Ca2+ channel.75 Recently, however, it has been noted that L type Ca2+ channels may contribute to the generation of the intermittent dystonia observed in these mice. Even more, the susceptibility of L type channels to voltage dependent facilitation may promote the abnormal motor phenotype in thetg mice.95
Acknowledgments
I would like to thank Drs A Darszon, T Nishigaki, and C L Treviño for helpful comments on the manuscript, and two anonymous reviewers for constructive criticism. Work in the author's laboratory is supported by the National Council for Science and Technology (Conacyt, Mexico) Grant 31735-N.