Article Text

Abstract
Kufor-Rakeb syndrome is an autosomal recessive nigro-striatal-pallidal-pyramidal neurodegeneration. The onset is in the teenage years with clinical features of Parkinson's disease plus spasticity, supranuclear upgaze paresis, and dementia. Brain scans show atrophy of the globus pallidus and pyramids and, later, widespread cerebral atrophy. We report linkage in Kufor-Rakeb syndrome to a 9 cM region of chromosome 1p36 delineated by the markers D1S436 and D1S2843, with a maximum multipoint lod score of 3.6.
- Kufor-Rakeb syndrome
- autozygosity mapping
- Parkinson's disease
- chromosome 1p36
Statistics from Altmetric.com
Parkinson's disease is a relatively common neurodegenerative disorder affecting approximately 1% of the population over the age of 50 years.1 The aetiology is complex and currently undetermined but involves loss of the dopaminergic neurones of the nigro-striatal tract.2 3 Since there is no known curative therapy, any Mendelian condition that manifests even some symptoms of idiopathic Parkinson's disease is of potential interest since insights into the perturbed neurophysiology found in such rare conditions may lead to novel therapies applicable to patients with the idiopathic form of the disease. We report our studies of a consanguineous Jordanian family with four members affected by Kufor-Rakeb syndrome, a neurodegenerative disorder involving the nigro-striatal tract, the globus pallidus, and the pyramids.
The Kufor-Rakeb syndrome was originally reported by Al-Dinet al 4 where they described clinical findings in five affected offspring of a consanguineous couple. These subjects showed a number of the features seen in idiopathic Parkinson's disease including a mask-like face, rigidity, and bradykinesia but the onset of the condition was much earlier, between 11 and 16 years of age, and the disease was rapidly progressive. Quoting the original paper: “The features of paralysis agitans were remarkable in their rate of development rendering the patients bed-ridden within less than two years of onset”. Therapy with levadopa resulted in significant improvement of extrapyramidal function for up to two years. Interestingly, intention tremor, a classical Parkinson's disease feature, was not seen. Additional disease features found in Kufor-Rakeb syndrome that are absent in idiopathic Parkinson's disease were spasticity resulting from corticospinal tract degeneration, a supranuclear upgaze paresis, and the development of dementia in all affected subjects. Although the Kufor-Rakeb syndrome has similarities to pallido-pyramidal syndrome, it seems to be distinguished as a separate entity by the presence of dementia and upgaze paresis seen in this family together with the lack of intention tremor.4-7 The oldest affected subject, who had a severe spastic paraplegia, was aphasic and demented, has died following a respiratory infection. It was not possible to perform a necropsy. MRI brain scans of affected family members showed the progression of disease with initial atrophy of the globus pallidus and pyramids, and later generalised brain atrophy.4
The inheritance pattern of Kufor-Rakeb syndrome was considered to be autosomal recessive as the parents had none of the symptoms seen in their children, they were second cousins, five of their nine offspring had the condition, and, of these, one was female with a phenotype indistinguishable from that of her four affected brothers. We therefore used autozygosity mapping to find the locus for Kufor-Rakeb syndrome with samples from the parents, four of their affected children, and one of their unaffected children. The “unaffected” subject was a 23 year old female; she had no abnormal neurological symptoms or signs when examined by the authors and was considered to have lived through the age of onset of the condition seen in her affected sibs.
The CHLC/Weber Human Screening Set version 8 (Research Genetics Inc) was used in a genome wide search. This panel contains 365 autosomal microsatellite repeat markers spaced at approximately 10 cM intervals with an average heterozygosity of 0.76. PCR amplification of all markers was performed according to the manufacturer's specifications using a Roboseq 4200 (MWG BioTech Ltd). Amplified markers were pooled and electrophoresed on an ABI Prism 377 gene sequencer (Applied Biosystems) with 4.2% polyacrylamide gels at 3000 V and 52°C for 2.5 hours. Fragment length analysis was undertaken using the ABI Prism Genescan and Genotyper 1.1.1 analysis packages.
Box 1
The allele frequencies for some of the markers we used, D1S228, D1S436, D1S1592, D1S2826, D1S2644, D1S199, and D1S2828, have not been published. These are required to generate lod scores from linkage analysis programs. Rather than estimate the allele frequencies, we sought a data source that had used these markers and had generated human data. CEPH has used these markers to genotype their CEPH reference families. By accessing http://www.cephb.fr/cephdb/dumps the data can be retrieved. Therefore, we could use the CEPH data to give us allele frequencies for markers D1S228, D1S436, D1S1592, D1S2826, D1S2644, D1S199, and D1S2828. We had to ensure, however, that our allele calling/identification was the same as that of CEPH. To do this we genotyped one of their reference samples, DNA from subject 1347-02, against the markers. This allowed us to ensure that we and CEPH had identical allele identification. We were then able to use the CEPH data to produce allele frequencies for our markers.
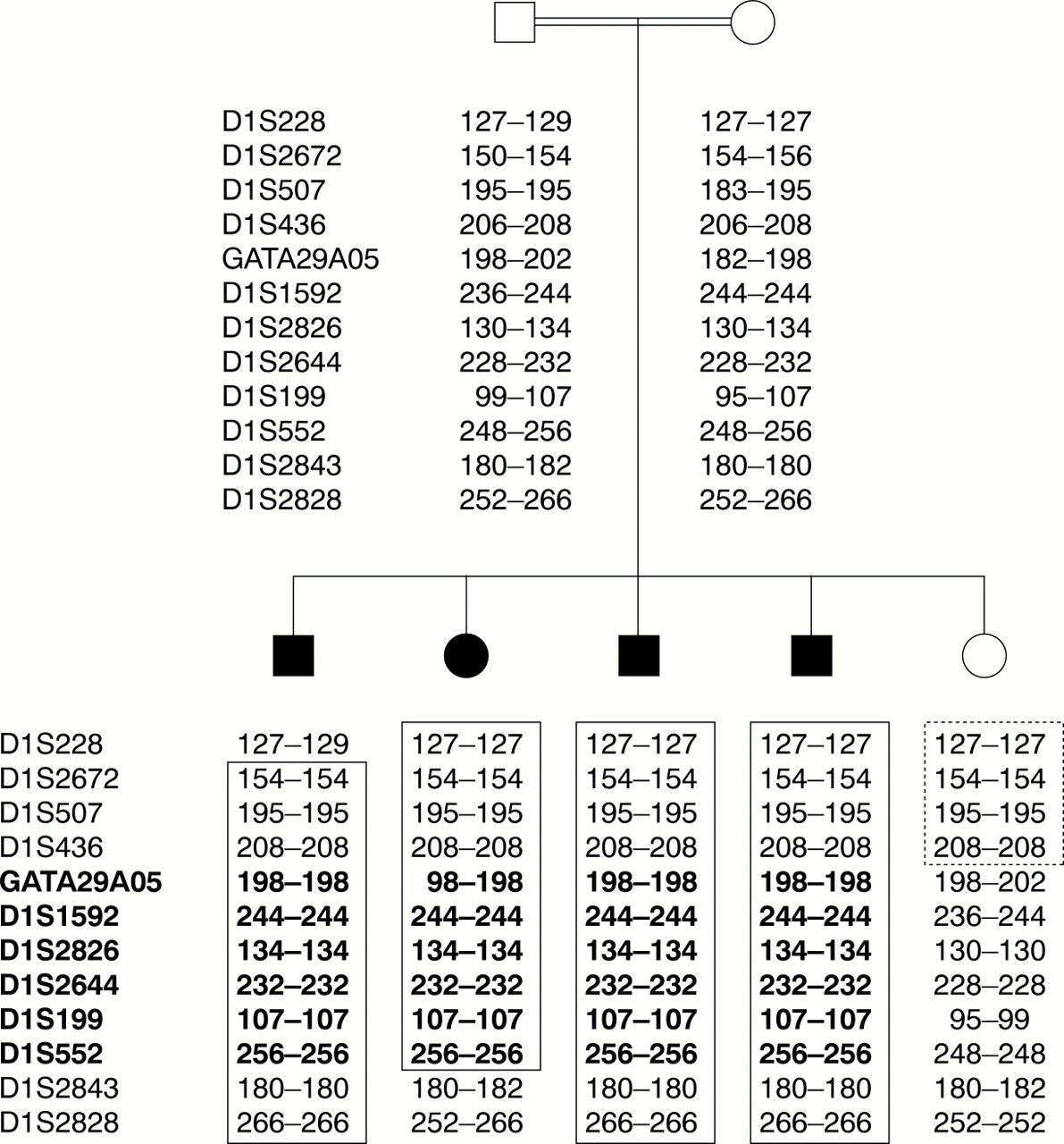
The initial genome search identified only one common region of homozygosity in all four affected subjects on chromosome 1p incorporating markers GATA29A05 and D1S552. There was no other region where two or more markers were homozygous or where there was a potential common haplotype involving three or more adjacent markers. Fine mapping was conducted by selecting markers from the ABI Linkage Mapping Panel Version I (Applied Biosystems), the Todd Panel,8 and the Marshfield Linkage Maps: tel - D1S228 - D1S2672 - D1S507 - D1S436 - (GATA29A05) - D1S1592 - D1S2826 - D1S2644 - D1S199 - (D1S552) - D1S2843 - D1S2828 - cen. This placed the region on chromosome 1 at band p36, with meiotic crossovers between markers D1S228-D1S2672 and D1S552-D1S2843, with D1S228 and D1S2843 defining the telomeric and centromeric boundaries. The results from the unaffected sib further refined the minimal critical region with a telomeric crossover between D1S436 and GATA29A05. Taken together, these results suggest that the region containing the Kuffor-Rakeb syndrome gene is approximately 9 cM in size with D1S436 and D1S2843 defining the telomeric and centromeric boundaries (fig 1). Information regarding marker order and relative distances was obtained from the Marshfield Linkage Maps. The marker order obtained from the Marshfield Linkage Maps was in agreement with that derived from the draft human genome. An autosomal recessive mode of inheritance with 95% penetrance was assumed. The disease allele frequency was estimated at 1 in 150. Allele frequencies for markers D1S228, D1S436, D1S1592, D1S2826, D1S2644, D1S199, and D1S2828 were calculated from the CEPH data for subject 1347-02 (see box 1). Pedigree allele inconsistencies were identified using PedCheck.9 Two point analysis was performed using the LINKAGE analysis programs10 and multipoint lod scores were computed by means of the GENEHUNTER program, version 2.0 beta.11 The two point lod score at θ=0 for markers in the critical region using CEPH data for allele frequencies were −∞ for D1S228 and D1S436, +1.14 for D1S1592, +2.53 for D1S2826, +2.40 for D1S2644, +2.57 for D1S199, and −∞ for D1S2828. The highest multipoint lod score was 3.6 for the chromosome 1p36 region containing markers D1S1592, D1S2826, D1S2644, and D1S199 (fig 2).

Nuclear pedigree (cf Al-Din et al4) of the family with Kufor-Rakeb syndrome showing microsatellite marker genotypes at chromosome 1p36. Marker order is tel-D1S228-D1S2672-D1S507-D1S436-GATA29A05-D1S1592-D1S2826- D1S2644-D1S199-D1S552-D1S2843-D1S2828-cen. The affected subjects are shown as shaded. The boxed region indicates the homozygous regions in the affected subjects. The dotted box indicates the autozygous segment in the unaffected sib. The minimum autozygous segment for Kufor-Rakeb syndrome is shown in bold.

Multipoint linkage analysis of the Kufor-Rakeb family across the autozygous segment at 1p36 showing lod score results for markers D1S228, D1S436, D1S1592, D1S2826, D1S2644, D1S199, and D1S2828.
Ubiquitin specific protease 1 (USP1A) has been mapped cytogenetically to the Kufor-Rakeb region 1p32-3612 and is a potential candidate gene because of the involvement of the ubiquitin degradation pathway in the autosomal recessive Mendelian form of Parkinson's disease PARK2.13However, the gene seems excluded by radiation hybrid mapping of theUSP1 EST stSG41379 and examination of the draft human genome browser placing the gene at 1p22, outside the candidate region for Kufor-Rakeb syndrome.
The initial features of Kufor-Rakeb syndrome are mask-like face, rigidity, and bradykinesia, including a response to dopaminergic therapies. This strongly suggests that there is a progressive loss of dopaminergic neurones in the nigro-striatal pathway, as seen in idiopathic Parkinson's disease. The clinical finding of progressive spasticity and pyramid atrophy on brain scan show that Kufor-Rakeb syndrome also involves corticospinal tract degeneration. One feature of Parkinson's disease is absent, tremor. This may be explained by the pallidal degeneration seen on brain scans of affected members of the Kufor-Rakeb family, as pallidal ablation is used in advanced Parkinson's disease to reduce symptoms including tremor.14 Kufor-Rakeb syndrome is probably, therefore, a nigro-striatal-pallido-pyramidal degeneration. We have mapped this disorder to a 9 cM region of chromosome 1p36. Eventual identification of the gene causing Kufor-Rakeb syndrome will lead to a greater understanding of the complex functions and diseases of the basal ganglia in man.
Acknowledgments
Electronic database information. BLAST server for sequence comparison: (http://www.ncbi/nlm.nih.gov/blast). For genetic linkage maps: Center for Medical Genetics, (http://www.research.marshfieldclinic.org/genetics/). For expressed sequence tags and gene assignments: Human Gene Map '99, (http://www.ncbi.nlm.nih.gov/genemap99). For the draft human genome browser: http://genome.cse.ucsc.edu/goldenPath/septTracks. For CEPH allele data: http://www.cephb.fr/cephdb/dumps.
We thank the family for their assistance and the Wellcome Trust who funded this work. ER is funded by Action Research, YC by the UK Medical Research Council, and JB by the British Heart Foundation.

{kind=link}
{kind=link}
{kind=link}