Article Text

Abstract
There are currently two clinically and molecularly defined forms of myotonic dystrophy: (1) myotonic dystrophy type 1 (DM1), also known as ‘Steinert’s disease'; and (2) myotonic dystrophy type 2 (DM2), also known as proximal myotonic myopathy. DM1 and DM2 are progressive multisystem genetic disorders with several clinical and genetic features in common. DM1 is the most common form of adult onset muscular dystrophy whereas DM2 tends to have a milder phenotype with later onset of symptoms and is rarer than DM1. This review will focus on the clinical features, diagnosis and management of DM1 and DM2 and will briefly discuss the recent advances in the understanding of the molecular pathogenesis of these diseases with particular reference to new treatments using gene therapy.
- myotonic dystrophy
Statistics from Altmetric.com
Myotonic dystrophy type 1
Prevalence
Estimates of the prevalence of myotonic dystrophy type 1 (DM1) range from approximately 1:100 000 in some areas of Japan to approximately 1:10 000 in Iceland, with a European prevalence of 3–15 per 100 000.1 Founder effects may have increased the prevalence in specific regions, such as Quebec, where the incidence rises to 1 in 500.2
Genetics
DM1 is caused by an expansion of an unstable CTG trinucleotide repeat in the 3′ untranslated region (UTR) of the gene DMPK (myotonic dystrophy protein kinase) which codes for a myosin kinase expressed in skeletal muscle—‘myotonin protein kinase’. The gene is located on chromosome 19q13.3.3 4
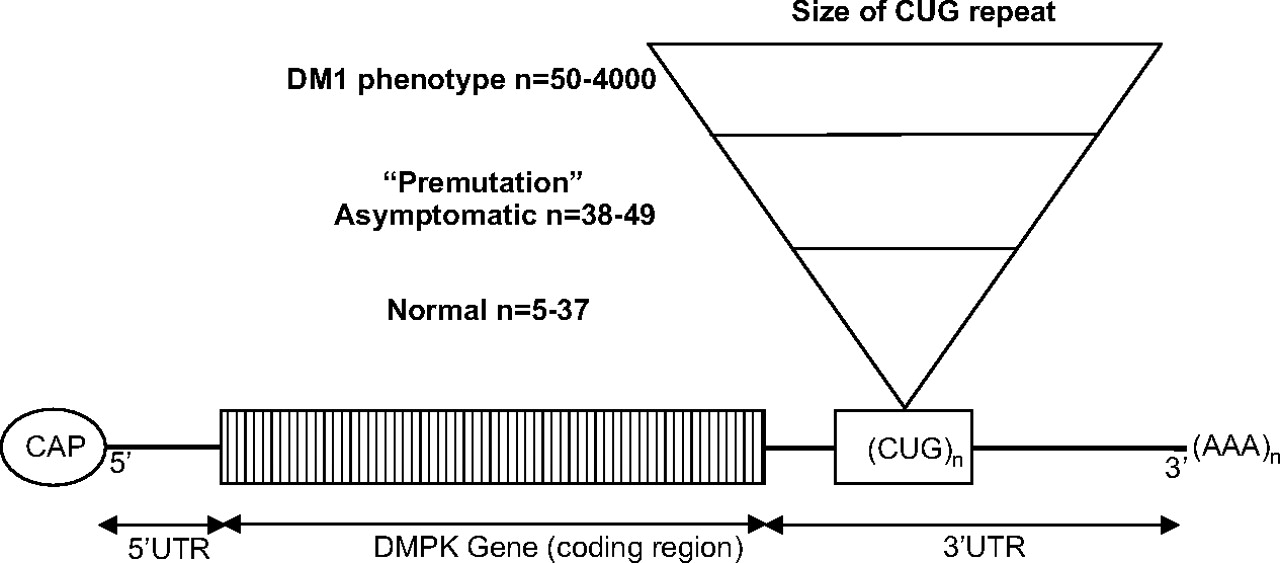
Normal individuals have between 5 and 37 CTG repeats (see figure 1). CTG repeat lengths exceeding 37 are abnormal. Patients with between 38 and 49 CTG repeats are asymptomatic but are at risk of having children with larger, pathologically expanded repeats.5 This is called a ‘pre-mutation’ allele. Full penetrance alleles occur with repeats greater than 50 CTGs and are nearly always associated with symptomatic disease although there are patients who have up to 60 repeats who are asymptomatic into old age and similarly patients with repeat sizes up to 500 who are asymptomatic into middle age. CTG repeat sizes in patients range from 50 to 4000. Molecular genetic testing detects mutations in 100% of affected individuals. Allele sizes were established by the Second International Myotonic Dystrophy Consortium (IDMC) in 1999.6 PCR analysis is used to detect repeat lengths less than 100 and Southern blot analysis to detect larger expansions. Predictive testing in asymptomatic relatives as well as prenatal and preimplantation diagnosis can also be performed.7

DMPK pre-mRNA with relationship between CUG repeat size and phenotype. DM1, myotonic dystrophy type 1; UTR, untranslated region.
Anticipation
DMPK alleles greater than 37 CTG repeats in length are unstable and may expand in length during meiosis and mitosis. Children of a parent with DM1 may inherit repeat lengths considerably longer than those present in the transmitting parent. This phenomenon causes ‘anticipation’, which is the occurrence of increasing disease severity and decreasing age of onset in successive generations. The presence of a larger repeat leads to earlier onset and more severe disease and causes the more severe phenotype of ‘congenital’ DM1 (figure 2).8 9 A child with congenital DM 1 almost always inherits the expanded mutant DMPK allele from their mother (figures 3–5), in contrast with other triplet repeat disorders where anticipation tends to occur through the paternal lineage, for example, Huntington’s disease. Paternal inheritance of congenital DM1 has been described but is very rare.10 Large CTG expansions are possibly toxic or negatively select against sperm compared with oocytes with larger expansions. To confuse the issue further, anticipation may be seen in patients with DM1 who inherit a smaller expanded CTG repeat from their father and expansions in these individuals tend to be greater than from mothers with a similar repeat size.11 12 The severity of anticipation is therefore dependent on the size of the repeat and the sex of the parent the expanded repeat is inherited from.13
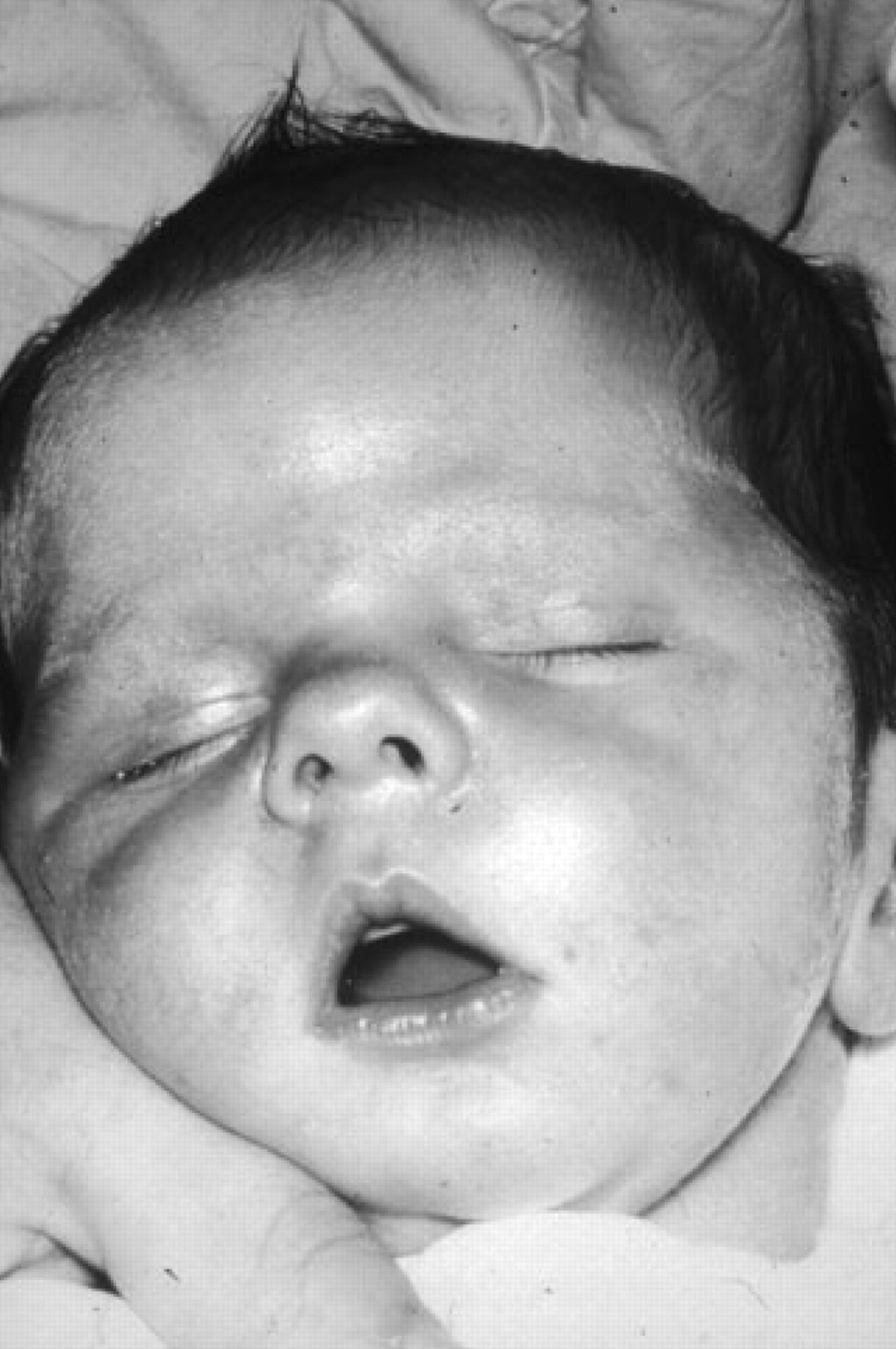
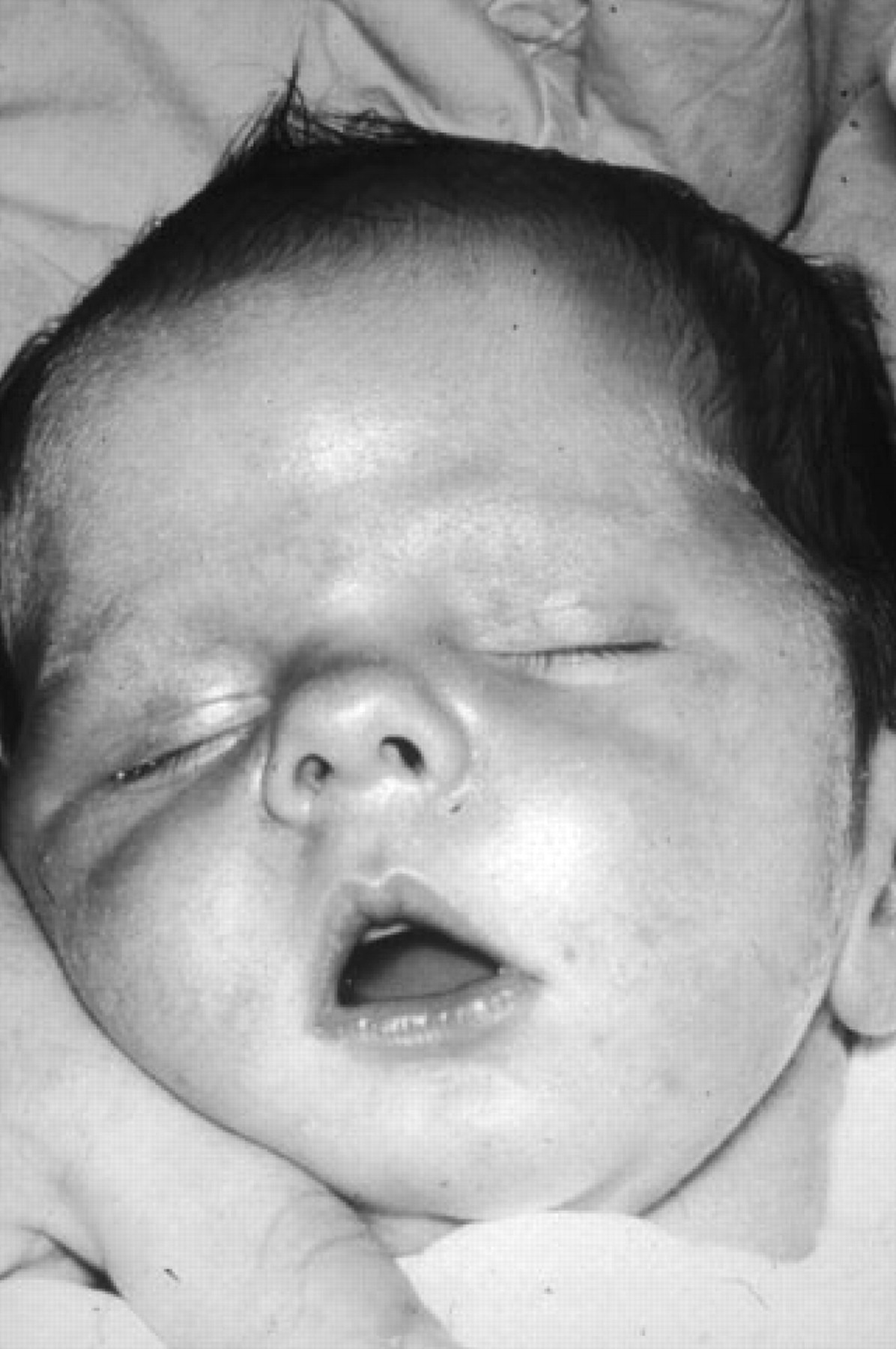
The typical tented upper lip (‘carp mouth’) appearance in congenital myotonic dystrophy.

Typical facial appearance of severe adult onset myotonic dystrophy showing weakness and atrophy of the facial muscles, wasting of the temporalis and ptosis (approximately 530 CTG repeats).

Patient is eldest son of patient in figure 3. He has congenital onset myotonic dystrophy and already shows similar features to his mother and frontal balding (approximately 1300 CTG repeats). Unfortunately, his younger brother (approximately 1500 CTG repeats) died suddenly and this was presumed to be secondary to a malignant cardiac arrhythmia.

A mother with DM1 in centre (approximately 230 CTG repeats) with her affected daughter on the left (approximately 630 CTG repeats) and affected son (approximately 730 CTG repeats). The son and daughter have expanded repeats compared with their mother due to anticipation through the maternal line.
Genotype–phenotype correlations
In general, longer CTG repeat expansions correlate with an earlier age of onset and more severe disease (table 1). CTG repeat size correlates more significantly with age of onset and disease severity below 400 CTG repeats.14 15 One explanation for the poor correlation of phenotype with repeat size above 400 repeats is that the DMPK CTG trinucleotide repeat length is mitotically unstable in individuals with DM1. This instability leads to somatic mosaicism for the size of the CTG expansion.16 The correlation between CTG repeat size observed in one tissue (eg, blood) often does not match the severity of the disease and the CTG repeat size in other organs (eg, muscle). Repeat size is often stable in some postnatal tissues (eg, blood) but not in others (eg, skeletal and cardiac muscle). Somatic instability occurs in mitotic and post-mitotic tissues, suggesting it is caused by changes in DNA repair mechanisms. There is no correlation between the size of the CTG repeats in muscle and the degree of weakness. In clinical practice, the CTG expansion is measured in blood and there is no additional clinical advantage of measuring repeat size in muscle.
Summary of the clinical findings, phenotype and CTG repeat length in myotonic dystrophy type 1
Non-genetic testing
The diagnosis of DM1 is made from DNA testing in an individual who is clinically suspected to have DM1. Non-molecular testing has been used in the past to establish the diagnosis of DM1 but now has no role in making the diagnosis in current practice. Patients may have had an EMG and occasionally a muscle biopsy and other tests before the diagnosis was clinically suspected. It is therefore valuable to be aware of the features of these investigations in patients with DM1. It should be emphasised that there is no clinical indication for performing a muscle biopsy to make the diagnosis of DM1. If the clinical features suggest DM1, but DM1genetic testing is negative, then DM2 testing should be performed.
Electromyography was the most helpful laboratory study before genetics were available. The combination of myotonic discharges and myopathic appearing motor units, predominantly in distal muscles and the face, is highly suspicious of DM1. The characteristic myotonic discharges occur as bursts of repetitive potentials on insertion of the needle. The potentials vary in both amplitude and frequency, and when played over a loudspeaker they resemble the sound of a diving propeller airplane and are called ‘dive bomber’ or ‘motorcycle’ potentials. Electrical myotonic discharges are not usually seen during infancy but fast runs of single fibre discharges approaching the pattern of myotonic discharges are suggestive of DM1.
Serum creatine kinase concentration may be mildly elevated in patients with DM1 but it is often normal in asymptomatic individuals.
Muscle biopsy is histologically grossly abnormal in clinically affected patients. Features include variability in fibre size, fibrosis, rows of internal nuclei, ring fibres (orientation of myofibrils at 90° to the fibre), sarcoplasmic masses, early type I fibre atrophy and increased numbers of intrafusal muscle fibres.
Some patients demonstrate low levels of immunoglobulin G.
Liver function tests are elevated in up to 50% of patients of unknown aetiology
Brain MRI often shows cerebral atrophy, increased white matter signals on T2 weighted images and thickening of the cranial vault.17
Clinical features
Patients with DM1 can be divided into four main subtypes based on their clinical presentation. Table 1 summarises these subtypes.
Congenital DM1
Congenital DM1 is not a severe early form of ‘classical’ DM1 but a distinct clinical phenotype with distinct clinical features (figure 2). Congenital DM1 often presents before birth as polyhydramnios and reduced fetal movements. After delivery, the main features are severe generalised weakness, hypotonia and respiratory compromise. Affected infants have an inverted V-shaped (also termed ‘tented’ or ‘fish-shaped’) upper lip, which is characteristic of severe facial weakness and makes suckling difficult. Mortality from respiratory failure is high. Failure to thrive, club feet and feeding difficulties are common problems but surviving infants experience gradual improvement in motor function, can swallow and independently ventilate. Cognitive and motor milestones are nevertheless delayed and all patients with congenital DM1 develop learning difficulties and require special needs schooling. Cerebral atrophy and ventricular enlargement are often present at birth.18 19 Children with congenital DM1 are able to walk. Faecal soiling can be problematic. A progressive myopathy and the other features seen in the classical form of DM1 can develop although this does not start until early adulthood and usually progresses slowly.20 Patients often develop severe problems from cardiorespiratory complications in their third and fourth decades.
Childhood onset DM1
The diagnosis of childhood DM1 is often missed in affected adolescents or children due to the lack of neurological problems and apparently negative family history.21 In contrast with the congenital DM1 patients, the sex of the parent does not influence the development of childhood onset DM1. Facial weakness is common but without the characteristic ‘tented’ upper lip appearance of the congenital form. Dysarthria and hand muscle myotonia may be prominent features with retarded motor development. Low intelligence and other psychosocial problems are often the principal management problem.22 There is increasing evidence of early conduction abnormalities and from the age of 10 annual electrocardiograms and consideration of electrophysiological studies should be a part of routine management.
‘Classical’ adult onset DM1
Muscular dystrophy
The predominant symptom in classic DM 1 is distal muscle weakness, leading to difficulty with performing tasks requiring fine dexterity of the hands and foot drop, particularly affecting ankle dorsiflexors. The characteristic facies is caused by weakness and wasting of the facial, levator palpebrae and masticatory muscles giving rise to ptosis and the typical myopathic or ‘hatchet’ appearance (figures 3–5). The neck flexors and finger/wrist flexors are also commonly involved. Ophthalmoplegia is described but is rare. Muscle weakness progresses slowly. An axonal peripheral neuropathy may add to the weakness.23
Myotonia
Myotonia may interfere with daily activities such as using tools, household equipment or doorknobs. Handgrip myotonia and strength may improve with repeated contractions—‘warm up phenomenon’.28 The warm up phenomenon can also improve speech production.29 Myotonia can be typically elicited by percussion of the thenar eminence with a tendon hammer—‘percussion myotonia’. Maintained contraction of the eyes or hands—‘eye closure’ and ‘handgrip’ myotonia—are less commonly elicited than percusion myotonia.
Ocular
Posterior subcapsular cataracts develop in most patients at some time in their illness.30 Some patients will have a history of cataracts at an early age without any other symptoms and then may develop muscle symptoms later in their disease.
Cardiac
Conduction disturbances and tachyarrhythmias are common in DM1 and contribute significantly to the morbidity and mortality of the disease.31–34 The cardiac histopathology demonstrates fibrosis, particularly of the conducting system and sinoatrial node, myocyte hypertrophy and fatty infiltration.35 EM demonstrates prominent I-bands and myofibrillar degeneration.
In one 10 year follow-up study by Mathieu et al26 there was a 7.3 times greater mortality in DM1 than the general population. Mean age of death was 53 years. There was a positive correlation between age of onset and age at death. Thirty per cent of the deaths were due to cardiac complications and 40% were due to respiratory complications. The cardiac abnormalities included sudden unexpected death, presumed to be due to a malignant arrhythmia, progressive left ventricular dysfunction and ischaemic heart disease although there is no conclusive evidence of early atherosclerosis in DM1. Tachyarrhythmias, bradyarrhythmias and conduction abnormalities were common, including a prolonged PR (40%) interval, a wide QRS (25%), sustained and paroxysmal atrial fibrillation (25%) as well as monomorphic and polymorphic ventricular tachycardia and ventricular fibrillation.
Groh et al36 investigated sudden death in patients with DM1 and found that a severe abnormality on the ECG (defined as a prolonged PR and/or QRS, second/third degree heart block or a non-sinus rhythm) and a diagnosis of an atrial tachyarrhythmia predicted sudden death. Approximately 50% of patients with sudden death in whom an electrodiagnosis could be made had a ventricular tachyarrhythmia. Over a 5.7 year follow-up, 7% of patients died from sudden death and 8% from respiratory failure.
Conduction defects and arrhythmias can occur at an early age when the myopathy is relatively mild and correlates poorly with CTG repeat expansion size.31 There may therefore be a role for genetic screening of asymptomatic children of affected patients. Physical activity precipitates arrhythmia and therefore exercise testing with ECG is sometimes recommended in young patients with DM1 before intensive sports are undertaken.37
In contrast with cardiac arrhythmias, cardiomyopathy is uncommonly a major clinical concern for patients.38
Central nervous system
Minor intellectual deficits are present in many patients, in contrast with congenital and childhood onset DM1 in which cognitive milestones are delayed and psychosocial issues are a greater clinical problem. The intelligence of patients with classical DM1 may be incorrectly assumed to be reduced because of facial expression, speech disturbance and apathy. Age related cognitive decline has been reported in some adults.39 40 Avoidant, obsessive–compulsive and passive–aggressive personality features have also been reported.41 42 Nocturnal apnoeic episodes are a common manifestation and are due to obstructive apnoea and primary central apnoea. Daytime sleepiness is extremely common and may be related to loss of serotoninergic neurons in the dorsal raphe and superior central nucleus of the brainstem.43–45 Patients have an abnormal central ventilatory response without the usual hyperpnoea produced by an increasing carbon dioxide concentration. This is associated with an abnormal sensitivity to barbiturates, morphine and other drugs that depress the ventilatory drive contributing to postoperative respiratory complications.
Gastrointestinal tract
Cholecystitis and symptoms referable to gallbladder function are frequent. Gallstones occur as a result of increased tone of the gallbladder sphincter, and cholecystectomy has a high morbidity due to postoperative respiratory complications. Liver function tests are often elevated for unknown reasons.46
Decreased peristalsis in the hypopharynx and the proximal oesophagus leading to dysphagic symptoms are common but uncommonly lead to parenteral feeding. Aspiration pneumonia is very common and the contribution of dysphagia, in the context of neuromuscular respiratory failure, results in high levels of morbidity and mortality from pneumonia. Patients often complain of constipation and may develop pseudo-obstruction and megacolon which may be due to either myotonia or smooth muscle loss in the bowel. Slow gastric emptying (28%), diarrhoea (50%) and occasional faecal incontinence (30%) occur in patients. The commonest gastrointestinal symptom complex is similar to irritable bowel syndrome. In 28% of patients, gastrointestinal problems occurred before the diagnosis of DM1 was made and 25% of patients find the worst symptoms are gastrointestinal related. The burden for patients of gastrointestinal related symptoms has generally been underecognised.47 48
Endocrinopathy
Endocrine abnormalities include disturbances of the thyroid, pancreas, hypothalamus and gonads. Testicular atrophy, with the disappearance of the seminiferous tubules, leads to infertility in men. Infertility may occur in otherwise asymptomatic patients.49 In women, habitual abortion and menstrual irregularities are common. Although diabetes mellitus is no more common in DM1 than in the general population, a glucose tolerance test is often associated with abnormally high glucose levels, particularly at 3 h into the test, suggesting insulin insensitivity. An associated overproduction of insulin is due to insulin resistance secondary to abnormal splicing of the insulin receptor mRNA to a more insulin insensitive isoform.50–52
Skin
Patients, more often males than females, commonly develop early frontal balding. Pilomatrixomata and epitheliomas can occur, especially on the scalp, and can be confused with sebaceous cysts.53 54
Respiratory
Weakness and/or myotonia of the diaphragm and a susceptibility to aspiration from swallowing problems increase the risk of respiratory compromise and aspiration pneumonia, usually in individuals with advanced disease and swallowing difficulties.55
Psychiatry
Anxiety and depression are common, and quality of life can be seriously impaired.56 Patients often have apathy which may be confused with significant depression.44 57
Pregnancy
Women with DM1 are at risk of complications during pregnancy, including increased spontaneous abortion rate, prolonged labour, retained placenta and postpartum haemorrhage.58 59
General anaesthesia
General anaesthesia is associated with significant complications in DM1. These are usually respiratory. Mathieu et al60 found a 10% risk of postoperative complications due to general anaesthesia, including prolonged respiratory depression from anaesthetic agents and postoperative pneumonia, especially following cholecystectomy.61
Prognosis of classical DM1
Several studies have evaluated lifespan and mortality in DM1 (table 1).25 26 The most common causes of death were pneumonia/respiratory failure, cardiovascular disease, sudden death/arrhythmia and neoplasms. Die-Smulders et al25 found that 50% of adult onset DM1 patients were either partially or totally wheelchair bound shortly before death.
Asymptomatic/late onset DM1
Arsenault et al62 investigated 102 DM1 patients with between 50 and 200 CTG repeats and found that most patients with between 50 and 99 were asymptomatic apart from 38% who had cataracts. Myotonia, weakness and excessive daytime sleepiness were far more common in patients with between 100 and 200 CTG repeats. Some patients with between 50 and 100 CTG repeats can nevertheless develop severe DM1 and some patients with between 300 and 500 CTG repeats can be asymptomatic. The most useful reason for detecting the DM1 mutation in this group of patients is often to identify other affected family members and to enable genetic counselling.
Neuropathology
Neurons in the limbic system and/or brainstem contain tau associated neurofibrillary tangles in DM1 and DM2, suggesting a common neuropathological process and a possible link with the CNS features of DM1, including apathy and sleepiness.63 64
Myotonic dystrophy type 2
DM2 was previously termed proximal myotonic myopathy and shares many of the features of DM1.65–68
DM2 is is an autosomal dominant disorder caused by a mutation in the ZNF9 gene on chromosome 3q21. ZNF9, the gene encoding zinc finger protein 9, is the only gene known to be associated with DM2. The first intron in ZNF9 contains a complex repeat motif (TG)n(TCTG)n(CCTG)n. Expansion of the CCTG repeat causes DM2.69 70The repeat expansion for DM2 is much larger than for DM1, ranging from 75 to over 11000 repeats. Unlike DM1, the size of the repeated DNA expansion does not correlate with age of onset or disease severity in DM2. Anticipation is less evident clinically in DM2. A congenital form of DM2 has not been reported.
Clinical features of DM2
DM2 is a multisystem disorder characterised by myotonia (90%) and muscle weakness (82%). The onset of DM2 is typically in the third decade, with the most common presenting symptom being muscle weakness although myotonia during the first decade has been reported.65 66 DM2 patients commonly have prominent muscle pain, stiffness and fatigue in comparison with DM1 (table 2) although muscle pain may be underestimated in DM1. The weakness typically affects proximal muscles, including the neck, elbow extension and hip flexors in comparison with early DM1 which initially tends to affect distal upper limb muscles.65
Comparative features of myotonic dystrophy type 1 (DM1) and type 2 (DM2) (adapted from Washington Neuromuscular website http://neuromuscular.wustl.edu/)
Other clinical features include cardiac conduction defects (19%), posterior subcapsular cataracts (36–78%, increasing with age) and endocrine changes, including insulin insensitivity (25–75%, increasing with age) and testicular failure (29–65%). Diabetes mellitus type 2 may be more common in DM2 than in DM1. Cognitive manifestations in DM2 include problems with organisation, concentration and word finding, and excessive daytime sleepiness. Conduction abnormalities are more common in DM1 than DM2 but cardiac screening is still necessary as the absolute cardiac risk in DM2 is not fully understood. Table 2 compares the main features of DM1 and DM2.
Prognosis of DM2
DM2 is a clinically milder disease than DM1. DM2 patients less commonly require assistive devices (canes, walkers, wheelchairs, scooters) than those with DM1 although they experience increasing difficulties climbing stairs as the disease progresses.
Molecular basis of DM1 and DM2
The mutation in DM1 occurs in an untranslated but transcribed portion of the 3′ UTR of the DMPK gene. There have been three postulated pathological molecular mechanisms in DM1. It was initially suggested that the mutant DMPK allele could cause a reduction in normal DMPK allele function and therefore the DM1 phenotype would be caused by haploinsufficiency. However, there is little evidence that there is a reduction in myotonin protein kinase levels or dysfunction of the protein. This led to the suggestion that the effect of the DMPK mutation may alter the expression of the adjacent genes, in particular DMWD and SIX5 (figure 6). However, there is increasing evidence that the transcribed DMPK pre-mRNA is directly toxic and results in abnormal splicing of other mRNA transcripts, including those of the muscle chloride ion channel, CLCN1.71 This may occur by the accumulation of splicing factors within ribonuclear accumulations of mutant DMPK or by a more direct effect of mutant DMPK on splicing factors within the nucleus, independent of the ribonuclear inclusions. A similar mechanism has been proposed for DM2. Figure 6 demonstrates the proposed molecular effects of altered splicing factors (CUG-BP and MBNL1) by mutant pre-mRNA in DM1 and 2. Splicing factors are required to convert pre-mRNA into mRNA, and increased CUGBP and decreased MBNL1 in DM1 possibly causes a reversion to more fetal mRNA species.72 The loss of several adult mRNA species causes defects in diverse tissues and leads to the multisystem nature of DM1 and DM2.73–75

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Summary of the molecular pathogenesis of myotonic dystrophy type 1 (DM1) and myotonic dystrophy type 2 (DM2), a ‘spliceopathy’. Adapted from Gatchel et al.82
Other myotonic disorders
There have been no further genetic causes of multisystem myotonic dystrophies identified although patients with DM1/2 gene negative myotonic dystrophies have been described. The IDMC has agreed that any newly identified multisystem myotonic dystrophies will be sequentially named as forms of myotonic dystrophy. Le Ber et al76 suggested that a family had DM3 but this was subsequently demonstrated to be an unusual presentation of inclusion body myopathy with Paget's disease and frontotemporal dementia77 caused by mutations in valosin containing protein.
Other hereditary disorders associated with myotonia are myotonia congenita, caused by mutations in CLCN1, paramyotonia congenita and its variants caused by mutations in SCN4A and hyperkalaemic periodic paralysis, also caused by mutations in SCN4A. These diseases do not have a prominent dystrophic component and are therefore often referred to as the ‘non-dystrophic myotonias’.
Management
Even though there is currently no cure for myotonic dystrophy, the active management of patients involves monitoring expected complications of the disease. Cardiorespiratory disorders are responsible for 70% of the mortality in DM1 and many of these patients could have been treated by active monitoring and a lower threshold for input. There is very little specific treatment that is distinct for DM2 and the multisystem pathologies of DM2 are similarly treated and monitored.
Muscle weakness
Dehydroepiandrosterone (DHEA)
DM1 may be associated with low circulating dehydroepiandrosterone (DHEA) levels.78 79 An initial pilot study suggested that DHEA may improve myotonia and muscle strength in DM1.80 A recently reported multicentre, randomised, double blind, placebo controlled trial was performed in France, involving 75 ambulatory adults with DM1 who received an oral replacement dose (100 mg/day), a pharmacological dose (400 mg/day) of DHEA or placebo. The primary endpoint was the relative change in the manual muscle testing score from baseline to week 12. There was no evidence that a 12 week treatment with replacement or pharmacological doses of dehydroepiandrosterone improved muscle power.81
Insulin-like growth factor 1 (IGF1)
Mecasermin rinfabate (SomatoKine/iPLEX) is a combination of recombinant insulin-like growth factor 1 (IGF1) and its binding protein, BP-3 (rhIGF-1/rhIGFBP-3). IPLEX was approved in the USA in December 2005 for the treatment of children with growth failure due to severe primary IGF-1 deficiency (primary IGFD). There have been early studies demonstrating that iPLEX may improve insulin insensitivity and function of DM1 myoblasts.83 84 An early phase 2 safety and tolerability study of daily subcutaneous iPLEX has suggested some promising early results with an increase in muscle mass in DM1 patients as well as improvements in other systems such as gastrointestinal function and lipids. The trial is not recruiting and is in the process of completion. (http://clinicaltrials.gov/ct2/show/NCT00233519?term=Muscular+Dystrophy&rank=46).
Creatine
Phosphocreatine has the potential to buffer the intracellular stores of ATP within muscle fibres. A recent Cochrane review of creatine treatment in all muscle diseases came out in favour of creatine in the treatment of ‘dystrophies’. The assessment included six trials of ‘dystrophies’, including two of DM1 and one of DM2 as well as three of dystrophinopathies. Unfortunately, the two trials of DM1 demonstrated mixed results, with one demonstrating a tendency to benefit with an increase in maximum voluntary contraction (MVC) of 8.4%85 and the other trial demonstrated slight harm with a reduction of −3.1% in MVC86 with an overall non-significant effect of Z=0.66 and p=0.51.87 Shneider-Gold and colleagues88 found a small but significant benefit (an 8.8% increase in MVC) of creatine in patients with DM2. The trials were crossover in design, carried out over a short period (8–12 weeks of treatment phases) and included small numbers of patients (68 patients in total for DM1 and 10 patients for DM2). Further assessment of the effects of creatine in larger double blind randomised trials may be warranted and routine use of creatine in DM1 cannot be currently recommended.
Exercise training
Strength training or aerobic exercise programmes may maximise muscle and cardiorespiratory function and prevent additional disuse atrophy in patients with muscle disease.89 However, over exerting may cause more rapid disease progression.
One randomised trial compared the effect of strength training versus no training in 36 patients with DM1. There were no significant differences between training and non-training groups for the primary or secondary outcome measures.90 Even though this does not prove the benefit of aerobic exercise in DM1, it would suggest that, contrary to previous thinking, moderate intensity strength training appears not to do any harm in DM1 and should be encouraged in all patients.
Myotonia
The main drugs that have undergone clinical trials for myotonia are sodium channel blockers (mexiletine, phenytoin, procainmide), tricyclic antidepressive drugs, benzodiazepines, calcium antagonists, taurine and prednisolone, and have been the subject of a Cochrane review.91 Ten randomised controlled trials have assessed the effectiveness of 12 different drug treatments. In total, 143 patients were treated, of whom 113 had myotonic dystrophy and 30 had myotonia congenita. Two small crossover studies, without a washout period, demonstrated a significant effect of imipramine and taurine in DM1. One small crossover study with a washout period demonstrated a significant effect of clomipramine in myotonic dystrophy. The studies were all too small to enable a meta-analysis to be performed.
Myotonia is often not a major symptom in patients with DM1 and many patients decline treatment especially when offered a potentially arrhythmogenic drug.
Cardiac arrhythmias
Tachyarrhythmia and conduction block may be responsible for up to 30% of fatalities in DM1.26 Early insertion of pacemakers does prevent some sudden cardiac deaths, but not all, suggesting that many patients may die from malignant tachyarrhythmias. It is extremely common to find conduction abnormalities on a resting ECG but electrophysiological studies (EPS) are probably required to assess the risk of developing bradyarrhythmias and tachyarrhythmias and the need for an implantable cardioverter defibrillator and/or permanent pacemaker. Eighteen per cent of patients with a normal 24 h Holter monitor have inducible ventricular tachycardia in the absence of symptoms on EPS.92 It is not certain how many of these patients develop dangerous arrhythmias and require insertion of an implantable cardioverter defibrillator. The RAMYD Study is a prospective multicentre Italian study to evaluate the arrhythmic risk in 540 DM1 patients and hopefully this will provide some answers to the uncertainties surrounding risk stratification of cardiac disease in DM1.
Respiratory
Sleep disordered breathing may be caused by central apnoea and/or obstructive sleep apnoea and may contribute to patient morbidity, especially excessive daytime sleepiness. All patients with excessive fatigue/sleepiness require an overnight sleep study as a minimum investigation. If sleep disordered breathing is significant then non-invasive ventilation should be offered to the patient although it is often not well tolerated. If the sleep study is normal, or if symptoms persist in spite of treatment of sleep disordered breathing, then CNS stimulant drugs (eg, modafinil, dexamphetamine or methylphenidate) can be used. Modafinil (200–400 mg/day) has undergone most investigation, and three crossover studies93–95 and one open label study96 have all found benefit to variable degrees.
A Cochrane review of psychostimulants in DM1 found the current level of evidence ‘inconclusive’ and suggested further trials were needed.97 There are clearly some patients, perhaps even an undefined subset, who from clinical experience respond to modafinil. We would therefore recommend modafinil at 100 mg/day, increased in 2 weeks to 100 mg twice daily. If this fails to help then the dose can be doubled and the patient reassessed in 2 months. If there is still no clear benefit then the drug should be stopped. Theoretical risks of cardiac and psychiatric side effects have not been borne out in clinical trials in DM1 or for other disorders such as narcolepsy. There have been some rare cases of severe skin eruptions although these are rare. Headache and gastrointestinal upset tend to be the commonest side effects.
Gastrointestinal
There have been no randomised trials examining the role and treatment of dysphagia in DM1. Many patients will not complain of dysphagia even when there is significant aspiration on video fluoroscopy. The patient's quality of life compared with the relative risk of aspiration often needs to be assessed. Many patients tend to take a conservative approach which consists of dietary manipulation (thickening their food and adding supplements) and adopting safer swallowing techniques.98 Parenteral feeding is not common in patients in the UK although this should be discussed if the patient has significant aspiration or recurrent chest infections due to recurrent aspiration.
Cholestyramine improves diarrhoea, incontinence and pain in most patients, possibly by preventing large bowel osmotic diarrhoea due to failure of absorption of bile salts in the terminal ileum. Norfloxacin may be effective when cholestyramine fails by treating bacterial overgrowth, and erythromycin may help slow gastric emptying by compensating for reduced motilin levels in DM1.47 48
Cataracts
Cataract formation occurs early in DM1 and can significantly worsen balance which is already impaired by power loss. Patients should be aware of the need for early reporting of worsening vision so that a prompt referral for potential cataract surgery can be made.
Endocrine
Even though the incidence of overt diabetes is probably no greater in DM1 than in the general population, patients tend to have impaired glucose tolerance. The clinical effects of this on patient prognosis are uncertain.
Therapy services
Physiotherapy may not only help with the management of foot drop and gait by using ankle foot orthoses and programmed exercises but also with the treatment of respiratory failure and the encouragement of exercise. Occupational therapy may help hand function but wrist splints are poorly tolerated by patients. The most important role of occupational therapy in DM1 is with home adaptations and the assessment of safety. Speech and language therapy is often helpful in assessing the degree of aspiration and discussing treatment options for dysphagia and aspiration. A clinical nurse specialist with a specific interest in muscle disease often acts as a link with the main hospital and provides consistency in monitoring the patient. Social workers are vital for housing and social support, especially towards the severe stages of the disease.
Patient care and organisations
Patient held care cards have been implemented in many centres and may improve compliance with monitoring the disease. In one study, 79% of cardholders had a recommended ECG within the last year compared with 3.5% of non-cardholders. In the UK, the main charitable organisations are the Myotonic Dystrophy Support Group (MDSG) (http://www.mdsguk.org/news.htm) and the Muscular Dystrophy Campaign (MDC) (http://www.muscular-dystrophy.org/). These organisations provide a significant support network for patients and carers as well as funding for research and treatment.
Recommended screening protocol
The protocol below is by no means all inclusive and has little evidence based research to support it but is the minimum standard of care for patients with DM1 that we would suggest.
Yearly ECG with a low threshold for cardiology referral for consideration of EPS and/or REVEAL device insertion if there is marked prolongation of PR interval or QRS complex, the development of any form of conduction block or if the patient develops cardiac symptoms such as palpitations or syncope.
Sleep study—any symptoms of excessive daytime sleepiness or obstructive sleep apnoea need investigating. Vital capacity measurement may be helpful at predicting patients developing neuromuscular respiratory failure.
Fasting glucose, if suspicion of diabetes.
Yearly assessment for cataracts by neurologist—patient reporting of worsening vision is also important.
Access to therapy services/exercise programme.
Consider psychostimulants for excessive daytime sleepiness.
Discuss prenatal testing and genetic counselling of family members if required.
Discuss gastrointestinal symptoms and management.
Discuss treatment options for myotonia if required although often not needed.
Discuss Care Card, patient support and future treatments.
Gene therapy
Recent advances in our understanding of the underlying molecular mechanisms involved in myotonic dystrophy has generated new approaches for more specific and effective treatments for DM1 and DM2. The development of targeted molecular treatments, especially antisense therapy, has achieved great success in vitro and in animal models, although the translation of this to human trials has understandably lagged behind. The main contributions to these achievements will be discussed.
Langlois et al99 were one of the first groups to demonstrate hammerhead ribozyme (a form of post-transcriptional silencing distinct from antisense therapy) mediated destruction of ribonuclear inclusions in myotonic dystrophy myoblasts, associated with a preferential reduction in mutant DMPK mRNA.
Furling et al100 demonstrated a reversal of the abnormal phenotype in DM1 myoblasts by preferential reduction of mutant DMPK, using a retroviral vector to produce a 149 bp antisense RNA complementary to 13 CUG repeats and to the 110 bp region following the CUG repeat sequence in DMPK 3′ UTR. This was one of the first studies to show a reversal of cellular phenotype.
Kanadia et al101 102 demonstrated the reversal of RNA missplicing and myotonia after overexpression of MBNL1 in a mouse model of DM1. Overexpression of MBNL1 in vivo was achieved by injection of recombinant adeno associated viral vector into the skeletal muscle of the transgenic mice. This study was one of the first to demonstrate an improvement in phenotype after the onset of symptoms and pathology in an animal.
Mahadevan et al103 developed a conditional transgenic mouse model of DM1 by overexpression of a normal DMPK 3′ UTR. This recapitulated the clinical and pathological features of DM1 in the absence of RNA inclusions but with increased CUG-BP1 levels. By silencing the transgene, the phenotype dramatically improved and suggested that the molecular deficits in DM1 can be reversed although whether this can occur after decades of exposure to mutant DMPK in humans, in comparison with weeks in transgenic mice, remains unsubstantiated.
Altered splicing of the muscle specific chloride channel 1 (ClC-1) has been shown to cause the myotonic phenotype of DM1 and this has recently been shown to be reversible in mouse models using morpholino (a small molecule to alter gene expression) antisense oligonucleotide to modify splicing of ClC-1 mRNA.104
Conclusions
DM1 is the commonest cause of adult onset muscular dystrophy. There is currently no cure but effective management is likely to significantly reduce the morbidity and mortality of patients who have perhaps not received as much clinical attention as warranted. The enormous advances in the understanding of the molecular pathogenesis of DM1 and DM2 has resulted in a novel disease mechanism involved in some myodegenerative and possibly neurodegenerative disorders. These diseases have been called the ‘spliceopathies’ and are mediated by a primary disorder of RNA rather than proteins. This has heralded the use of gene therapy which has already produced significant disease modifying effects in vitro and in animal models. The dawn of gene therapy for DM1 and DM2 appears to be very close and the near future is an exciting time for clinicians and patients alike.
References
Footnotes
Competing interests None.
Patient consent Obtained.
Provenance and peer review Commissioned; externally peer reviewed.