Article Text

Statistics from Altmetric.com
Hereditary progressive dystonia is characterised by lower limb dystonia of childhood onset with marked diurnal fluctuation and shows a dramatic and stable response to low dose levodopa. The disease is transmitted in autosomal dominant inheritance, and Segawa et al 1 proposed hereditary progressive dystonia as a new disease entity in the early 1970s. Dopa responsive dystonia, which was first proposed by Nygaard et al 2 in 1988, is essentially identical to hereditary progressive dystonia although it may include some other heterogeneous dystonias. The GTP cyclohydrolase I (GTP-CH I) gene on chromosome 14 is the causative gene of hereditary progressive/dopa responsive dystonia3 and more than 20 different mutations have been reported. We report a novel non-sense mutation in the GTP-CH I gene in a genetically confirmed sporadic Japanese patient.

{kind=link}
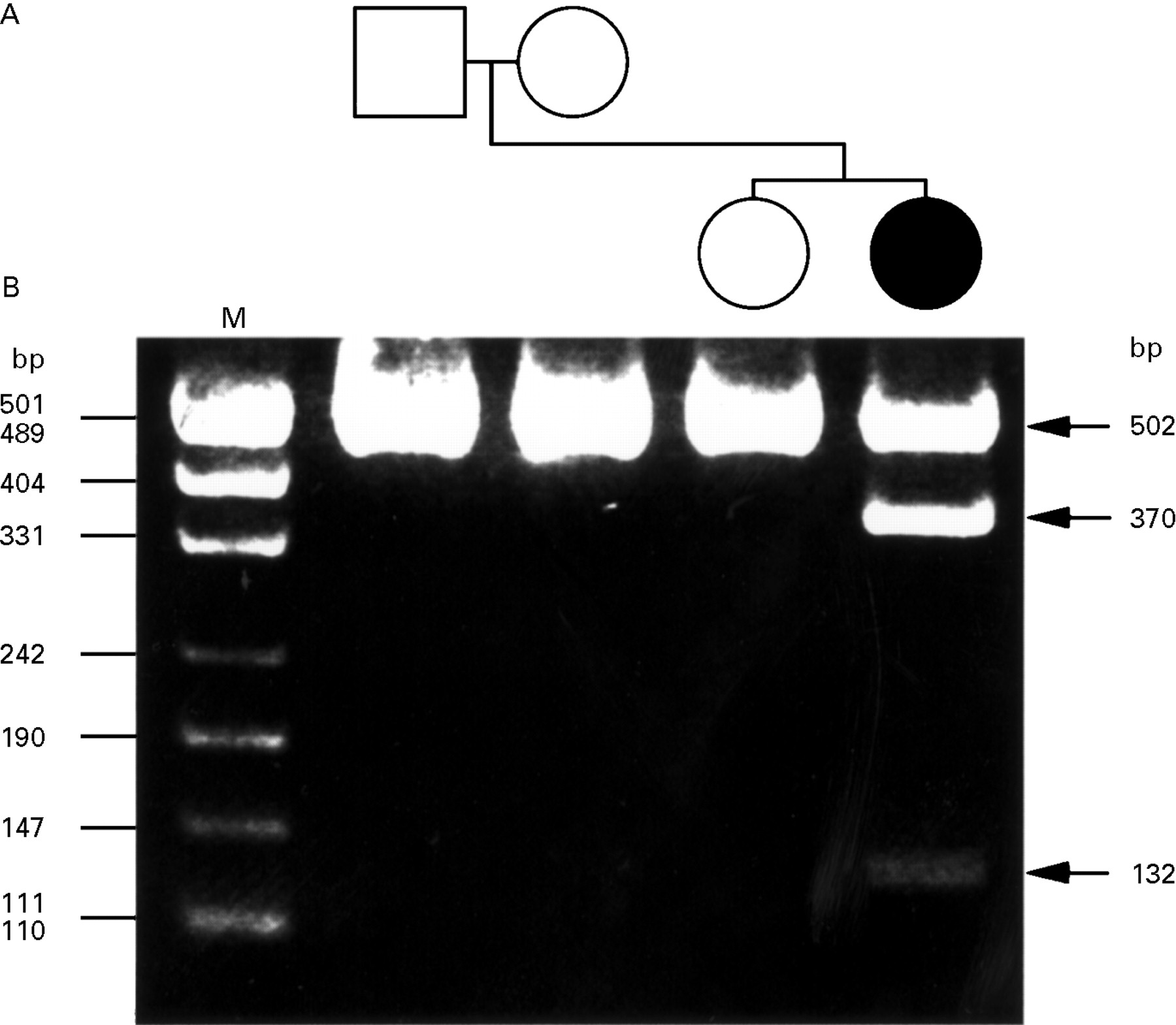
(A) Pedigree of a Japanese hereditary progressive/dopa responsive dystonia family with mutation in the GTP-CH I gene. The affected propositus is represented by solid symbol, and unaffected family members, by open symbols. (B) Agarose gel electrophoresis of Eco57I restriction pattern in the exon 1 of GTP-CH I gene. PCR product (502 bp) including exon 1 of the GTP-CH I gene from the patient was partially digested into two fragments of smaller sizes (370 and 132 bp) due to a Eco57I cleavage site created by a mutation in exon 1 of the gene. The 502 bp fragments from the unaffected family members remained undigested. Lane M=pUC19 DNA MspI digestion marker.
A 12 year old Japanese girl developed gait disturbance with a dystonic pes equinovarus posture in the right leg at the age of 4 years. These symptoms showed diurnal fluctuation; they were mild in the morning and worsened later in the day, and sleep improved the symptoms. They dramatically and continuously responded to low dose levodopa/carbidopa (100 mg/day) without adverse effects. Other members of her family, comprising the parents and one sister, had no symptoms.
Blood specimens of the patient and of the parents and sister were available for genetic analysis. For mutation analysis, genomic DNA was extracted from EDTA anticoagulated peripheral blood. Fragments of DNA containing the entire coding region of the GTP-CH I gene were obtained from genomic DNA by polymerase chain reaction (PCR) according to the method of Ichinose et al.3 Direct nucleotide sequencing of PCR products was performed with an automated DNA sequencer (Applied Biosystems 310) using the same primers as were used for amplification.
Direct nucleotide sequencing of genomic DNA of the patient showed a G to A transversion in exon 1 of the GTP-CH I gene (data not shown). This mutation produces a substitution of the tryptophan residue (TGG) with a stop codon (TGA) at position 96, which creates a new Eco57I cleavage site. The sequencing of the parents and the sister of the patient showed no mutation. To confirm the mutation, exon 1 was amplified and digested by Eco57I in all the four subjects. The restriction fragment length polymorphism consisted of two fragments (370 and 132 bp) in the mutant allele and one fragment (502 bp) in the normal allele (figure). The restriction pattern in the patient was consistent with heterozygous status, consisting of one each of the mutant and normal alleles, whereas that of the parents and sister, with homozygous status of two normal alleles. No other mutations were detected in the coding region of the gene.
The patient presented with typical clinical features of hereditary progressive dystonia. A new mutation in the GTP-CH I gene of the present patient causes a stop codon, and this mutation is most likely the pathogenic mutation. GTP-CH I catalyses the initial and rate limiting steps of tetrahydrobiopterin synthesis. Tetrahydrobiopterin is an essential cofactor for tyrosine hydroxylase, the rate limiting enzyme in the dopamine synthesis pathway. GTP-CH I activity in patients with hereditary progressive dystonia is less than 20% of that in normal subjects.3 The non-sense mutation in exon 1 confirmed in the present patient would have caused premature truncation of the GTP-CH I protein with a loss of an estimated 60% of the amino acid residues from the C terminal, and GTP-CH I activity, though it was not measured, may have been reduced below the critical threshold. To date, 22 different mutations in the GTP-CH I gene have been identified worldwide, and there seems to be no evidence of the founder effect.
To our knowledge, only a single case (patient 2 of Furukawa et al 4) was confirmed to be genetically sporadic. They identified the G to A transversion at the splice acceptor site of intron 1 in the GTP-CH I gene, causing skipping of the entire exon 2 in the mature mRNA. In the present family, the mutation was confirmed in the propositus only, and not in the parents and the sister. The restriction fragment length polymorphism generated by Eco57I consisted of one fragment (502 bp) common to the parents and sister and an additional two fragments (370 and 132 bp) unique to the patient. The present patient was thus confirmed to be genetically sporadic and heterozygotic, and is the second report of de novo mutation in the GTP CH-I gene.
Acknowledgments
We thank Dr Hiroki Takano, Department of Neurology, Brain Research Institute, Niigata University, for his invaluable advice. This work was partly supported by grants in aid for scientific research on priority areas from the Ministry of Education, Science and Culture, and from the Research Committee of Health and Welfare of Japan.