Article Text

Abstract
Objective To validate sphingomyelin (SM) dosage in the cerebrospinal fluid (CSF) of patients affected by chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) and Guillain-Barré syndrome (GBS) as a reliably assessable biomarker.
Methods We prospectively enrolled 184 patients from six Italian referral centres, in whom CSF SM levels were quantified by a fluorescence-based assay optimised and patented in our laboratory.
Results We confirmed increased levels of SM in the CSF of patients affected by typical CIDP (n=35), atypical CIDP (n=18) and acute inflammatory demyelinating polyradiculoneuropathy, AIDP (n=12) compared with patients affected by non-demyelinating neurological diseases, used as controls (n=85) (p<0.0001, p=0.0065 and p<0.0001, respectively). In patients with CIDP classified for disease stage, SM was higher in active CIDP compared with both controls and stable CIDP (p<0.0001), applying for a selective tool to treatment tailoring or withdrawal. SM was also increased in AIDP compared with axonal GBS, discerning the demyelinating from axonal variant of the disease. SM did not correlate with CSF protein levels, stratifying patients independently from commonly used CSF indexes, and displaying high specificity to avoid potential misdiagnosis. Finally, SM correlated with the main clinical scores and some neurophysiological parameters in patients with CIDP and AIDP.
Conclusions CSF SM is a diagnostic and staging wet biomarker for acquired demyelinating neuropathies and may effectively improve the management of patients affected by GBS and CIDP.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) and Guillain-Barré syndrome (GBS) are acquired immune-mediated neuropathies, characterised by strong heterogeneity in terms of clinical manifestations, prognosis and response to treatment. They are rare but treatable diseases and early recognition is essential.1
Diagnosis is based on combined clinical, laboratory and nerve conduction investigations but misdiagnosis, especially for CIDP, is very common.2 3 The European Federation of Neurological Societies/Peripheral Nerve Society (EFNS/PNS) guidelines improved diagnostic accuracy for CIDP but are often underused during routine clinical practice.4 In particular, nerve conduction studies (NCS), the variable response of patients to treatment and the disputable role of cerebrospinal fluid (CSF) proteins, are responsible for incorrect diagnosis.5–7 These observations lead to the conclusion that there are no effective biological markers to diagnose and classify GBS, CIDP and their variants.8
CSF remains the most suitable biological fluid for biomarker discovery in neurological diseases.9
In acquired demyelinating neuropathies, the inflamed spinal nerve roots can release products of the underlying pathological process into the CSF.10
We have previously shown that sphingomyelin (SM), a myelin-enriched lipid, is able to monitor myelin loss and remodelling in the peripheral nervous system. Additionally, SM levels, measured by a reliable, simple, rapid and inexpensive assay, are increased in the CSF of patients affected by CIDP and GBS. Of note, SM levels in the serum of these patients do not change compared with controls, excluding at the same time the clinical relevance of SM dosage in this biological fluid for CIDP and GBS and a passive diffusion of SM from the periphery.11
Here we describe an Italian multicentre prospective study, conducted on a sizeable and carefully selected number of patients, to validate SM dosage as a novel CSF index to be used in the routine clinical practice.
Methods
Participants and study design
We prospectively enrolled 184 neurological patients from six Italian referral centres devoted to the management of immune-mediated neuropathies. Patients were consecutively enrolled in each centre after obtaining approval from the Local Ethics Committee, starting from November 2018 and until the end of November 2019. At entry, every patient underwent a neurological examination by an expert group of physicians. In addition, NCS were performed in the presence of suspected neuropathy.
At the enrolment in the study and within 1 week from their admission into the hospital, all patients were subjected to spinal tap and blood withdrawal as part of the routine testing, following the consensus recommendations of the Italian Association of Neuroimmunology for immunochemical CSF examination.12
One millilitre of CSF collected for the clinical practice was immediately stored at −80°C for SM testing, to preserve lipid and protein integrity over time.13
We assigned patients to three major cohorts including definite GBS, definite CIDP and other neurological diseases (OND), used as controls, according to published criteria.9 14–16
Suspected CIDP, not fulfilling EFNS/PNS diagnostic criteria, were not included in the control group but analysed as an independent group, also referred to as ‘no EFNS/PNS CIDP’.
Inclusion/exclusion criteria, patient cohorts’ characteristics, data collection on demographical, laboratory, clinical and neurophysiological features were detailed in online supplemental information.
Supplemental material
SM assay
SM dosage was performed by a fluorescence-based assay optimised in our laboratory (see online supplemental information for further details) (patent: 102015000078276).11
Statistical analyses
Data shown in tables were expressed as mean±SD and CI, unless otherwise specified. Results in figures 1 and 2 were presented as mean±SEM. Statistical difference between two groups was determined using the two-tailed Student’s t-test. Multiple group comparison was performed by one-way analysis of variance followed by the Holm-Sidak test. Correlation coefficient was estimated by the Spearman’s rank correlation test. We evaluated SM test performance in terms of accuracy using the receiver-operating characteristic (ROC) curve analysis. Statistical difference was considered to be significant when p<0.05. All statistical analysis was performed using the Graph Pad V.7.0 (Prism) software.
Standard protocol approvals, registrations and patient consents
Written informed consent to use CSF for SM dosage and to access to personal health information was obtained from all the patients involved in the study following the World Medical Association’s Declaration of Helsinki.
Results
Patient cohorts
A total of 184 neurological patients were enrolled in the study.
Eighty-four patients were enrolled with clinical suspect of CIDP; among them, 53 fulfilled the clinical and electrodiagnostic EFNS/PNS criteria and were further divided in 35 typical (66%) and 18 atypical (34%) CIDP.14 Atypical CIDP included distal acquired demyelinating symmetric neuropathy, Lewis-Sumner syndrome, pure motor and pure sensory CIDP.17–19 Thirty-one patients among the 84 were not confirmed as definite, probable or possible CIDP. These patients were analysed as the independent group ‘no EFNS/PNS CIDP’. The group included patients finally diagnosed as neuropathy of unknown aetiology (n=11), axonal neuropathy with different comorbidities including diabetes (n=8), hereditary neuropathy with pressure palsies (HNPP) (n=2), plexopathy (n=3), multifocal motor neuropathy (n=2), anti-myelin-associated glycoprotein neuropathy (n=1), amyotrophic lateral sclerosis (n=1), deficit of vitamin B12 (n=1) and two patients with undefined diagnosis.
Fifteen patients were diagnosed as definite GBS15 16; among them, NCS allowed to identify 12 acute inflammatory demyelinating polyradiculoneuropathy (AIDP) (75%) and 3 axonal GBS (25%).
Finally, 85 patients affected by non-demyelinating neurological disorders (also referred to as OND) were used as controls. This group included patients affected by non-immune-mediated axonal neuropathy (n=19), hydrocephalus (n=9), cerebrovascular disorders (n=9), cerebral tumour (n=1), cognitive impairment (n=11), plexopathy or spondylosis (n=6), cephalgia (n=4), muscle disorders (n=3), visual impairment (n=5), cerebellar syndrome (n=3), epilepsy (n=1) and 14 healthy patients.
Relevant demographic and immunochemical data on CSF and serum are summarised in table 1.
Demographic data and laboratory assessment
In table 2, clinical features including disease duration and activity, response to treatment and clinical scales are listed.
Clinical data of patients affected by CIDP and GBS
Motor and sensory electrophysiological findings in patients with CIDP and GBS are reported in online supplemental tables S1 and S2. Patients with just descriptive neurophysiology (n=7) were not included in these tables. We considered demyelinating features of NCS essential to corroborate CIDP and AIDP diagnosis.17 20
Supplemental material
Supplemental material
SM in the CSF of patient cohorts
We found that SM concentrations were higher in the CSF of patients affected by typical CIDP (n=35), atypical CIDP (n=18) and AIDP (n=12) compared with OND (n=85) (p<0.0001, p=0.0065 and p<0.0001, respectively) (figure 1A). These data confirmed the ability of SM testing to identify demyelination in the CSF of patients affected by acquired demyelinating neuropathies,11 although some overlap between demyelinating neuropathies and OND could be seen.
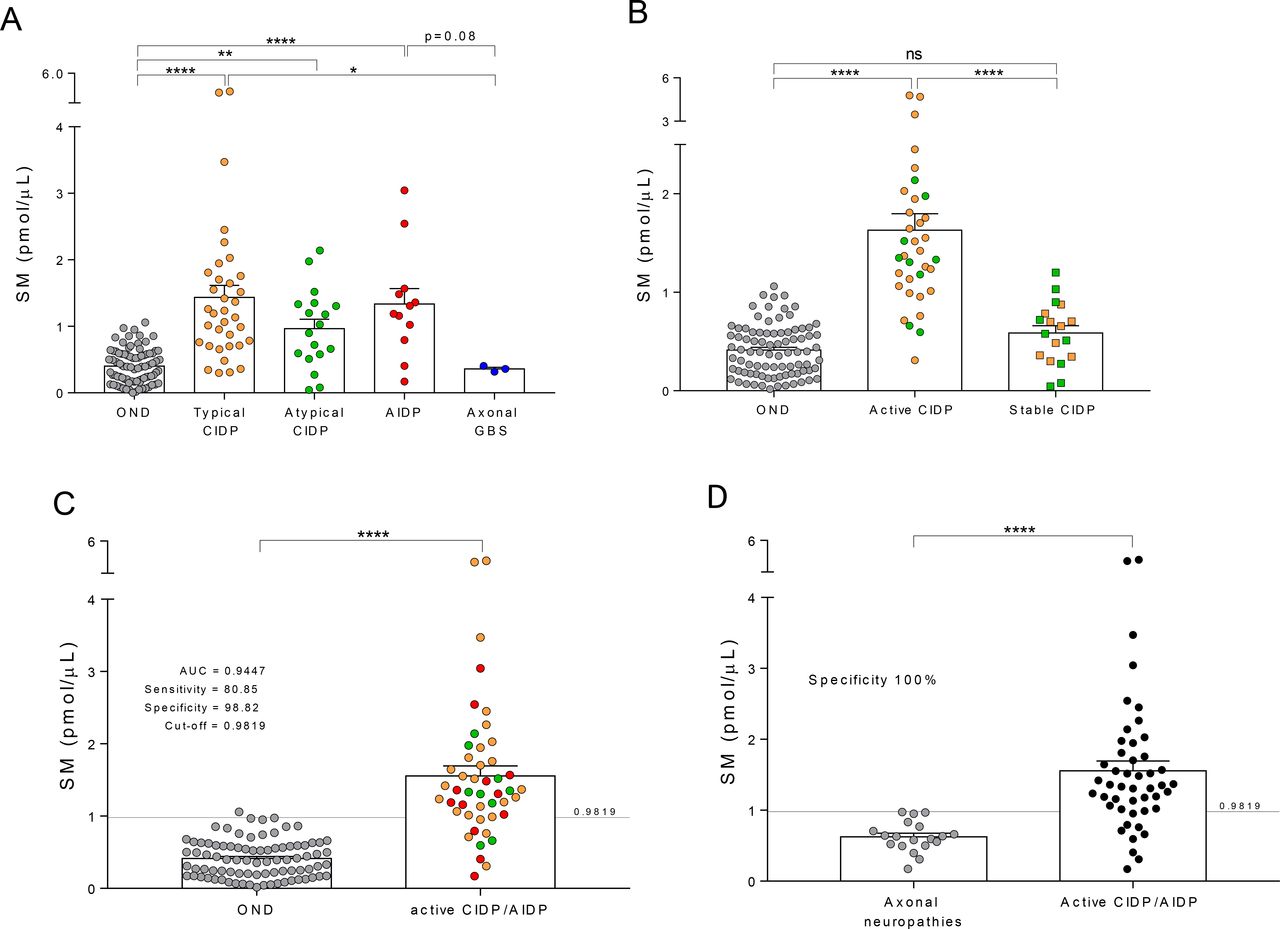
CSF SM levels in patients affected by CIDP and GBS. (A) Patients affected by acquired demyelinating neuropathies displayed increased levels of SM in the CSF, compared with patients affected by non-demyelinating diseases. In particular, patients with both typical and atypical CIDP showed increased levels of CSF SM compared with controls (ie, OND) (1.44±0.18 and 0.97±0.14 vs 0.41±0.03 pmol/µL). Consistently, SM levels in the CSF of patients with AIDP were increased compared with both OND and patients affected by axonal forms of GBS (1.34±0.23 vs 0.41±0.03 and 0.30±0.03 pmol/µL). (B) Patients with CIDP, independently from the clinical form, displayed increased levels of CSF SM when in the active stage of the disease compared with OND and stable CIDP (1.63±0.17 vs 0.41±0.03 and 0.59±0.07 pmol/µL); conversely, no difference in terms of SM content was found between clinically stable patients with CIDP and OND (0.59±0.07 vs 0.41±0.03 pmol/µL). (C) A ROC curve analysis was performed to define the characteristics of SM levels as a CSF biomarker of active demyelination. A cohort of patients affected by active CIDP and AIDP was compared with the OND cohort. SM levels significantly increased in patients compared with OND (1.56±0.14 vs 0.41±0.03 pmol/µL). We found that AUC for SM was 0.9447, indicative of a very good discriminatory biomarker. CSF SM testing exhibited high sensitivity (80.85%) and specificity (98.82%) in the identification of patients affected by chronic and acute demyelinating polyradiculoneuropathy. The SM cut-off for optimum sensitivity and specificity was 0.9819 pmol/µL. (D) SM testing displayed a 100% specificity in the identification of patients with CIDP in the active stage of the disease and patients with AIDP from a cohort of patients with axonal neuropathies (1.56±0.14 vs 0.63±0.05 pmol/µL). Data were presented as mean±SEM. Unpaired two-tailed t-test was used for statistical comparison between two groups. Holm-Sidak multiple comparison test after one-way analysis of variance was used for statistical comparison of multiple groups. *p<0.05; **p<0.01; ****p<0.0001; ns=not significant. Specificity and sensitivity of SM assay were tested by ROC curve analysis. A cut-off value for SM was also calculated. All statistical analysis was performed using the Graph Pad V.7.0 (Prism) software. AIDP, acute inflammatory demyelinating polyradiculoneuropathy; AUC, area under the curve; CIDP, chronic inflammatory demyelinating polyradiculoneuropathy; CSF, cerebrospinal fluid; GBS, Guillain-Barré syndrome; OND, other neurological diseases; ROC, receiver-operating characteristic; SM, sphingomyelin.
This overlap was much less evident when SM concentrations were compared in AIDP versus axonal GBS, which showed levels similar to those of the OND, despite the low number of patients with GBS (p=0.08 and p=0.88, respectively).
Since in this study we enrolled patients with CIDP displaying clinically heterogeneous features, we decided to define different subgroups, to appreciate the relevance of SM in the complex management of the disease. First, analysing patients with CIDP grouped based on the clinical form (ie, typical and atypical CIDP), we did not find any significant difference in the CSF content of SM; instead, we detected a strong heterogeneity of SM levels within each group (figure 1A). Then, for both typical and atypical CIDP we considered the stage of the disease, thereby dividing patients in stable and active. Active CIDP was defined for those patients displaying a clinical relapse documented by the change of at least one point in the clinical scales. Stable CIDP was defined for those patients displaying unvaried clinical scores. Based on that, we found that SM was higher in active CIDP (n=35) compared with both stable CIDP (n=18) and OND (n=85) (p<0.0001 and p<0.0001, respectively), whereas we did not observe difference between the stable CIDP and OND groups (p=0.22) (figure 1B and online supplemental figure S1A). Furthermore, while stable patients with CIDP were clinically unvaried, they displayed a residual inflammatory component that prevented their identification from the active CIDP by currently used laboratory tests, namely CSF to serum albumin concentration quotient (QAlb) (figure 1B and online supplemental figure S1B).
To further test the ability of SM to detect myelin breakdown, we also merged AIDP with the active CIDP group to form a larger cohort of patients (also referred to as active CIDP/AIDP) in which an ongoing demyelination process, at the moment of spinal tap, was deemed to be present. As expected, SM levels were higher in the CSF of these patients (n=47) compared with OND (n=85) (p<0.0001) (figure 1C).
ROC curve analysis performed on these patients’ cohorts defined CSF SM levels as a very good discriminatory biomarker (area under the curve=0.9447) with an optimum cut-off of 0.9819 pmol/µL (online supplemental figure S1C). This cut-off is definitely similar to the one we calculated in our previous study (1.18 pmol/µL).11 Given that in the present multicentre prospective study a sizeable number of well-characterised patients have been investigated, we consider these results a validation of our test.
In CIDP and GBS, inflammation at the blood–spinal nerve root barrier causes a reduction of the CSF flow, and a proportional increase of the total protein content in the CSF.21 As expected, in our patients affected by CIDP and AIDP we found correlation between these indexes (online supplemental figure S2, panel A). Instead, in the same patients, SM levels did not correlate with CSF protein concentration and poorly with CSF to serum IgG concentration quotient (QIgG) and QAlb, pointing to the potentiality of SM to distinguish demyelination from inflammation (online supplemental figure S2, panel B).
Interestingly, SM concentrations were also able to recognise non-immune-mediated axonal from demyelinating neuropathy (p<0.0001), with a 100% specificity (figure 1D).
SM and CSF protein
In the absence of a diagnostic biomarker, increased CSF proteins remain an important supportive criterion for CIDP.7 22 However, the subjective interpretation of mild or moderate CSF albuminocytologic dissociation often contributes to misdiagnosis.3 5 22 In fact, although total protein concentration in the CSF largely depends from its levels in the bloodstream, most clinical laboratories (85%) use an upper reference limit of 0.45 g/L to consider proteins a supportive criterion for CIDP.3 23
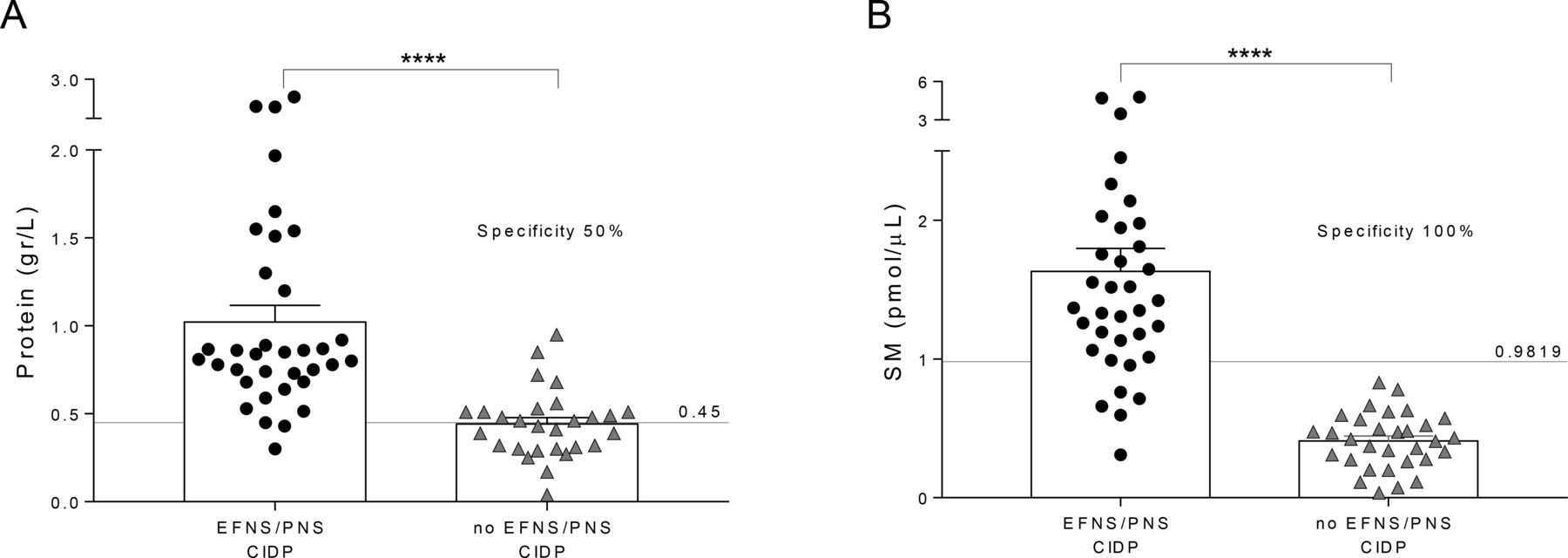
Among patients enrolled as suspected CIDP, 31 were not confirmed.14 In these patients, namely ‘no EFNS/PNS CIDP’, compared with patients affected by definite active CIDP (n=35), CSF protein concentration displayed just a 50% specificity to correctly identify patients with CIDP (figure 2A).3 23 Conversely, SM dosage in the CSF of the same patients demonstrated 100% specificity to correctly recognise those affected by definite CIDP, allowing to exclude patients affected by other neuropathies potentially misdiagnosed as CIDP. Actually, neither of our patients, with suspected CIDP but not fulfilling EFNS/PNS published criteria, had SM levels over the cut-off (figure 2B).
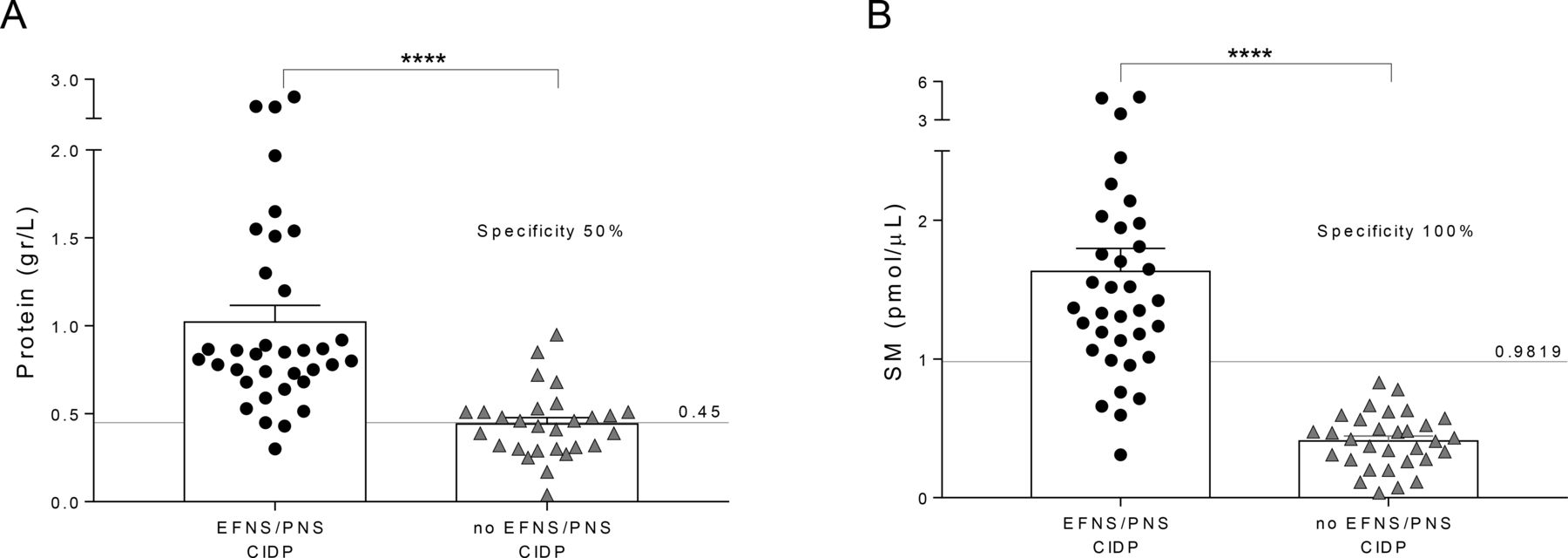
CSF protein and SM levels in potential misdiagnosed CIDP. (A) CSF protein concentration was tested for reliability in the identification of potential misdiagnosed patients with CIDP (also referred to as ‘no EFNS/PNS CIDP’) from patients affected by definite active CIDP (also referred to as ‘EFNS/PNS CIDP’). As expected, we found a significant increase of CSF protein in ‘EFNS/PNS CIDP’ compared with ‘no EFNS/PNS CIDP’ (1.02±0.09 vs 0.44±0.04 g/L). Using the URL of 0.45 g/L, CSF protein concentration displayed just a 50% specificity to correctly identify patients with CIDP. (B) SM levels in the CSF of the same patients showed a significant increase in ‘EFNS/PNS CIDP’ compared with ‘no EFNS/PNS CIDP’ (1.63±0.16 vs 0.41±0.04 pmol/µL). Of note, SM dosage demonstrated 100% specificity to correctly recognise the cohort affected by definite CIDP, allowing to exclude patients affected by other neuropathies and also potential misdiagnosed CIDP. Data were presented as mean±SEM. Unpaired two-tailed t-test was used for statistical comparison between two groups. ****p<0.0001. Specificity and sensitivity of CSF proteins and SM were tested by ROC curve analysis. All statistical analysis was performed using the Graph Pad V.7.0 (Prism) software. CIDP, chronic inflammatory demyelinating polyradiculoneuropathy; CSF, cerebrospinal fluid; EFNS, European Federation of Neurological Societies; PNS, Peripheral Nerve Society; ROC, receiver-operating characteristic; SM, sphingomyelin; URL, upper reference limit.
SM, clinical scores and neurophysiological parameters
Since in this study we enrolled patients displaying clinical heterogeneous pictures, we tested whether CSF SM was sensitive enough to mirror their degree of disability. In particular, we evaluated inflammatory neuropathy cause and treatment (INCAT), Overall Neuropathy Limitations Scale (ONLS) and INCAT sensory sum score (ISS) for patients affected by CIDP, and GBS disability scale and ONLS for patients affected by GBS; to grade muscle strength we evaluated Medical Research Council (MRC) sum score for both diseases.24–31 Correlation between these scales and CSF SM content was analysed in patients affected by active CIDP (n=35) (figure 3, panel A) and AIDP (n=12) (figure 3, panel B).

{kind=link}
{kind=link}
{kind=link}
Correlation of CSF SM levels with clinical scales in CIDP and AIDP. (Panel A) SM levels were correlated with clinical scales, including INCAT and ONLS to grade disease severity, and MRC sum score to grade muscle strength in the CSF of patients affected by active CIDP. Patients displayed a low but significant correlation of SM with all clinical scales. (Panel B) SM levels were correlated with clinical scales, including GBS disability scale and ONLS to grade disease severity, and MRC sum score to grade muscle strength in the CSF of patients affected by AIDP. Patients displayed a high and significant correlation of SM with GBS disability scale and a moderate but also significant correlation with ONLS and MRC sum score. Spearman’s rank correlation test was used for statistical analysis. *p<0.05; ***p<0.001. All statistical analysis was performed using the Graph Pad V.7.0 (Prism) software. AIDP, acute inflammatory demyelinating polyradiculoneuropathy; CIDP, chronic inflammatory demyelinating polyradiculoneuropathy; GBS, Guillain-Barrésyndrome; INCAT, inflammatory neuropathy cause and treatment; MRC, Medical Research Council; ONLS, Overall Neuropathy Limitations Scale; SM, sphingomyelin.
Patients with CIDP displayed a low but significant correlation of CSF SM levels with the majority of clinical scales (INCAT: r=0.357, p=0.0353; ONLS: r=0.3543, p=0.0368; MRC sum score: r=−0.4231, p=0.0113); in these patients we did not find any correlation of SM levels with ISS (data not shown).
Conversely, in patients affected by AIDP, we found a strong correlation of SM with GBS disability scale (r=0.8877, p=0.003) and a moderate correlation with ONLS and MRC sum score (r=0.5997, p=0.0426 and r=−0.606, p=0.0405, respectively). Indeed, GBS clinical scales are valid, sensitive, reliable and able to capture even subtle changes over time.24 32
Differently from SM, CSF protein concentration did not correlate with any clinical score, in the same cohorts of patients with CIDP and AIDP (online supplemental figure S3).
Supplemental material
We did not find any correlation between CSF SM and disease duration in both patients with CIDP and AIDP (data not shown).
Given that we collected neurophysiological data from four motor nerves and three sensory nerves for most of the patients enrolled in the study (online supplemental tables S1 and S2), we also correlated CSF SM levels with electrodiagnostic parameters in patients with active CIDP and AIDP. We found that SM levels correlated with proximal and distal compound motor action potential amplitude of the tibial nerve (r=−0.4474, p=0.0090; r=−0.4098, p=0.0179, respectively), with sensory nerve action potential amplitude of the median nerve (r=−0.6869, p=0.0329) and sensory conduction velocity of the ulnar nerve (r=−0.5889, p=0.0063) (online supplemental figures S4 and S5).
Supplemental material
Supplemental material
Discussion
This study provides evidence that CSF SM may represent a novel diagnostic and staging biomarker, useful for the early identification and classification of acquired demyelinating neuropathies in a clinical setting.
At present, effective treatments in clinical practice are available for most of these patients, but there is an unmet clinical need for objective biomarkers to improve diagnosis, stage patients, reliably assess disease progression, as well as evaluate therapeutic interventions. To this end, demyelinating polyradiculoneuropathy is a shared pathological hallmark in patients affected by CIDP and AIDP. While several molecules reflecting tissue damaging have been suggested as biomarkers, none of them is part of the routine clinical management of these patients so far.7 10 14
Treasuring the needs of CIDP/GBS community, we have been engaged for a few years in finding a wet biomarker that has to be endowed with clinical relevance, sensitivity and specificity, reliability, practicality and simplicity.33 34
In a retrospective study performed on patients affected by CIDP and AIDP, we found that CSF SM represents a promising candidate.11
To verify the issue, we planned the present study on a considerable number of clinically and neurophysiological well-characterised patients, prospectively enrolled. This allowed the exact classification of patients in definite subgroups corresponding to the most common variants of both CIDP and GBS, fundamental to confirm and eventually explore novel makings of SM dosage.
We confirmed the high sensitivity (80.85%) and specificity (98.82%) of SM dosage to identify patients affected by chronic and acute demyelinating polyradiculoneuropathy. In particular, we found higher levels of SM in the CSF of patients with CIDP affected by both typical and atypical form of the disease compared with controls. Among them, five patients displayed SM levels under the cut-off; interestingly, these patients had a moderate clinical impairment mainly characterised by a distal sensory involvement at the neurophysiological examination. Patients with AIDP also displayed increased CSF SM content compared with both controls and patients affected by axonal forms of GBS, although the low number of the latter ones. Overall, these results corroborated the diagnostic accuracy of SM testing.
Of note, a rapid and correct identification of demyelinating and axonal variants of GBS by CSF SM testing might help to overcome the current delay and possible misinterpretation of neurophysiology.15 35
The CSF albuminocytologic dissociation itself, that is considered a hallmark of GBS, does not discern between the different variants of the disease; in fact, rather it represents an index of inflammation and of reduced CSF flow rate and turnover caused by swellings in the area around the spinal roots.5–7 21 Instead, the SM testing, being the expression of myelin breakdown and remodelling more than of inflammation, may help to overcome the above limitation. To this end, inside the AIDP group, the two patients showing the lowest levels of SM were those enrolled in the early phase of the disease. It is likely that in these patients the inflammatory process had not yet caused substantial myelin damage.
Overall, due to these critical issues, increasing the number of patients with GBS analysed for CSF SM will be extremely important.
Concerning CIDP, we observed that clustering patients for disease activity allowed demonstrating that stable patients, independently from the clinical form and increased CSF inflammatory indexes, had SM levels comparable to those of the control cohort. Consistently, relapsing patients with CIDP showed a remarkable increase of SM in the CSF compared with both stable CIDP and controls. To this end, SM levels displayed to be more accurate than currently used CSF indexes to assign a patient with CIDP to the correct disease stage, and suggest a promising application in the evaluation of response to therapy.
Furthermore, an early and accurate diagnosis of CIDP is essential to engage a treatment and to prevent axonal damage, but is equally important to avoid severe side effects and associated costs, by prolonged therapy that needs to be stopped, when not necessary.26 36 In that sense, SM dosage may contribute to improve the pharmacovigilance of these patients.
We found that most patients with CIDP in the stable stage of the disease, based on SM levels, were treated (80%), but only 35% of them responded to therapy. Consistently, almost all patients with CIDP in the active stage displayed to be good responders to therapy (94%). SM testing facilitating the correct identification of disease stage, may therefore allow to distinguish patients who do not require further treatment either because cured, or become non-responders, from those still sensitive to therapy.
That CSF SM dosage may integrate into the management of CIDP and AIDP is further highlighted by the correlation with clinical scales, usually used to grade disease severity in these patients. Of interest, we found that this correlation was remarkable, especially in AIDP. This is expected, because of the acute nature of the disease, and the less likely existence of confounding factors in the clinical history, including disease duration, pharmacological treatment and comorbidities. Interestingly, also in patients with CIDP SM testing, although at a lower level of significance, correlated with clinical scores, in spite of the long-lasting disease duration and clinical heterogeneity.24
Given these promising results, SM might be envisaged as a CSF biological marker useful to corroborate ‘activity and participation level’of CIDP and AIDP in clinical trials.24
Notably, we did not observe any correlation between CSF protein and clinical scales in both CIDP and AIDP.
Leaders within the CIDP community also raised the issue about patients misdiagnosed as CIDP. This is a crucial unsolved aspect implying medical, social and economic consequences.19 37
The large percentage of misdiagnosed CIDP is linked to a poor utilisation of clinical and electrodiagnostic criteria developed by EFNS/PNS.36–38
Equally problematic is the CSF analysis that has the potential to confuse, rather than clarify the diagnosis.5–7 23
Of note, we found that SM assay displayed a 100% specificity in the correct identification of patients affected by CIDP and exclusion of potentially misdiagnosed patients. Instead, CSF protein index displayed just a 50% specificity.22 To this end, CSF SM levels may be a more specific and informative supportive criterion than CSF protein to correctly finalise a CIDP diagnosis. Interestingly, the ‘no EFNS/PNS CIDP’ group encompassed patients affected by different types of polyneuropathy including the demyelinating inherited form HNPP; all these patients commonly might be confused for CIDP owing to misleading clinical and neurophysiological features. Actually, SM assay displayed to be able to identify them overcoming this limit. Of course, future studies involving large cohorts of patients are necessary to definitely assess CSF SM performance on the topic.
Our study has shown that CSF SM measurement can achieve a diagnosis of immune-mediated demyelinating neuropathy in more than 80% of cases tested in a specialised clinical diagnostic setting. Hundred percent of the potentially misdiagnosed CIDP, whose identification would have been extremely laborious, are correctly identified. The vast majority of patients in the active stage of the disease are properly identified; given their remarkable clinical heterogeneity, we envisage that further studies taking into account age, disease duration and the effects of immunotherapy will be able to definitely legitimise the clinical relevance of CSF SM dosage in improving the management of these patients. Furthermore, due to the typical simplicity and practicality, CSF SM dosage may be clinically acceptable from a community in which subscribing to diagnostic guidelines is still a limitation.5
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors GC was responsible for study concept and design, data acquisition, analysis and interpretation, statistical analysis, obtaining funding and manuscript drafting. CD, MC, RF, ES, DS, DV, DF, FM, GS, AB, EC, PL, EN-O, AS and LB were responsible for patients’ enrolment, data acquisition, analysis and interpretation, and revision of the manuscript for intellectual content. LN was responsible for study concept and design, revision of the manuscript for intellectual content, manuscript drafting/revising and obtaining funding. All authors commented on the final version of the manuscript.
Funding GC is funded by the Associazione Italiana per lo studio del Sistema Nervoso Periferico (ASNP) and Associazione Italiana di Miologia (AIM) (Grant GMN, 2018). LN and LB are supported by the GSB/CIDP Foundation (Research Grant, 2018). LN is also supported by Fondazione Italiana Sclerosi Multipla (FISM, 2015/R/17) and by Institutional Research Funds from IRCCS Ospedale Policlinico San Martino.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The study was approved by local institutional ethical committees, following the approval obtained from our centre, as the study coordinator (CERLiguria P.R. 503REG2015 12 September 2018).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplemental information. All data relevant to the study are included in the manuscript or uploaded as supplemental information.Any additional data that may not be directly available will be shared by request from any qualified investigator from the corresponding author (Lucilla Nobbio—lnobbio@neurologia.unige.it; http://orcid.org/0000-0003-2150-3953).
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Linked Articles
- Editorial commentary